Ѫ�쵰��D�� ѪҺ��
Ѫ�쵰��D��
http://15130.dis.999120.net ��������
�����Ժ�ϸ������֢ ѪҺ��
�����Ժ�ϸ������֢������������Ѫ�쵰��
http://15131.dis.999120.net ��������
���ȶ�Ѫ�쵰�ײ� ѪҺ��
���ȶ�Ѫ�쵰�ײ������ȶ���Ѫ�쵰�ײ�
http://15132.dis.999120.net ��������
�ж��Ը���Ѫ�쵰��Ѫ֢ ѪҺ��
�ж��Ը���Ѫ�쵰��Ѫ֢
http://15133.dis.999120.net ��������
�Ŵ��Ը���Ѫ�쵰��Ѫ֢ ѪҺ��
�Ŵ��Ը���Ѫ�쵰��Ѫ֢
http://15134.dis.999120.net ��������
��Ѫ�쵰��Ѫ֢ ѪҺ��
��Ѫ�쵰��Ѫ֢����Ѫ�쵰��Ѫ֢
http://15135.dis.999120.net ��������
����˯����Ѫ�쵰���� ѪҺ��
����˯����Ѫ�쵰��������˯����Ѫ����������-�����ۺ���������ҹ��Ѫ�쵰��������ҹ��Ѫ�쵰����(����ƶѪ)������ҹ��Ѫ�쵰����֢
http://15136.dis.999120.net ��������
���財��ƶѪ ѪҺ��
���財��ƶѪ�����������ƶѪ
http://15137.dis.999120.net ��������
��Ѫ��ƶѪ ѪҺ��
��Ѫ��ƶѪ
http://15138.dis.999120.net ��������
������ABO��Ѫ֢ ѪҺ��
������ABO��Ѫ����ABOhemolyticdiseaseofnewborn��������ABO��Ѫ֢
http://15139.dis.999120.net ��������
RhѪ�Ͳ�����Ѫ�� ѪҺ��
RhѪ�Ͳ�����Ѫ��
http://15140.dis.999120.net ��������
�Ŵ������κ�ϸ������֢ ѪҺ��
�Ŵ������κ�ϸ������֢
http://15141.dis.999120.net ��������
�Ŵ�����Բ�κ�ϸ������֢ ѪҺ��
�Ŵ�����Բ�κ�ϸ������֢
http://15142.dis.999120.net ��������
���κ�ϸ������֢ ѪҺ��
���κ�ϸ������֢�����κ�ϸ������
http://15143.dis.999120.net ��������
��״ϸ��ƶѪ ѪҺ��
��״ϸ��ƶѪ����״ϸ����ƶѪ
http://15144.dis.999120.net ��������
�Ŵ��Ըɱ��ϸ������֢ ѪҺ��
�Ŵ��Ըɱ��ϸ������֢���Ŵ��Ըɱ�ϸ������֢
http://15145.dis.999120.net ��������
������-6-��������øȱ��֢ ѪҺ��
������-6-��������øȱ��֢
http://15146.dis.999120.net ��������
���5��-������ȱ��֢ ѪҺ��
���5��-������ȱ��֢
http://15147.dis.999120.net ��������
�����������칹øȱ��֢ ѪҺ��
�����������칹øȱ��֢�����������칹øȱ��֢
http://15148.dis.999120.net ��������
���Ǽ�øȱ��֢ ѪҺ��
���Ǽ�øȱ��֢�����Ǽ�øȱ��֢
http://15149.dis.999120.net ��������
��ͪ�ἤøȱ��֢ ѪҺ��
��ͪ�ἤøȱ��֢
http://15150.dis.999120.net ��������
�¿�����������������Ѫ��ƶѪ ѪҺ��
�¿�����������������Ѫ��ƶѪ
http://15151.dis.999120.net ��������
�俹����������������Ѫ��ƶѪ ѪҺ��
�俹����������������Ѫ��ƶѪ
http://15152.dis.999120.net ��������
���������ۺ��� ѪҺ��
���������ۺ��������������ۺ�֢
http://15153.dis.999120.net ��������
��������Ѫ�쵰���� ѪҺ��
��������Ѫ�쵰�������Ժ�����Ѫ�쵰������������Ѫ�쵰����֢
http://15154.dis.999120.net ��������
ҩ���������������Ѫ��ƶѪ ѪҺ��
ҩ���������������Ѫ��ƶѪ��ҩ����������Ѫ��ƶѪ��ҩ���շ�����������Ѫ��ƶѪ
http://15155.dis.999120.net ��������
��������Դ����Ѫ��ƶѪ ѪҺ��
��������Դ����Ѫ��ƶѪ����������������Ѫ��ƶѪ
http://15156.dis.999120.net ��������
�о���Ѫ�쵰���� ѪҺ��
�о���Ѫ�쵰����exercisehemoglobinuria��������Ѫ�쵰�����о���Ѫ�쵰����֢���˶���Ѫ�쵰����֢
http://15157.dis.999120.net ��������
Ѫ�ܲ�����Ѫ��ƶѪ ѪҺ��
Ѫ�ܲ�����Ѫ��ƶѪ��Ѫ������Ѫ��ƶѪ
http://15158.dis.999120.net ��������
�����������Ѫ��ƶѪ ѪҺ��
�����������Ѫ��ƶѪ
http://15159.dis.999120.net ��������
�黯���������Ѫ��ƶѪ ѪҺ��
�黯���������Ѫ��ƶѪ��AsH3�������Ѫ��ƶѪ
http://15160.dis.999120.net ��������
Ǧ�ж��������Ѫ��ƶѪ ѪҺ��
Ǧ�ж��������Ѫ��ƶѪ
http://15161.dis.999120.net ��������
ͭ�ж��������Ѫ��ƶѪ ѪҺ��
ͭ�ж��������Ѫ��ƶѪ
http://15162.dis.999120.net ��������
��Ѫ���ۺ��� ѪҺ��
��Ѫ��֢�ۺ�������Ѫ-��֢�ۺ�������Ѫ��֢�ۺ�֢����Ѫ���ۺ�������Ѫ�������ۺ���
http://15163.dis.999120.net ��������
�̷������ظμ����ļ���ϸ������֢ ѪҺ��
�̷������ظμ����ļ���ϸ������֢
http://15164.dis.999120.net ��������
�Ŵ��Է�߲���� ѪҺ��
�Ŵ��Է�߲�������Ŵ��Է�߲��֢
http://15165.dis.999120.net ��������
�����߲���� ѪҺ��
�����߲��������ɫ߲����
http://15166.dis.999120.net ��������
�ٷ���Ƥ��߲���� ѪҺ��
�ٷ���Ƥ��߲������������߲������֢״��߲����
http://15167.dis.999120.net ��������
��ϸ��������߲���� ѪҺ��
��ϸ��������߲������congenitalerythropoieticporphyria�������Թ�������߲�����������Ժ�ϸ��������߲�������Ŵ��Ժ�ϸ��������߲��֢��Gunther������ϸ������߲������ϸ����������߲����
http://15168.dis.999120.net ��������
ԭ߲���� ѪҺ��
ԭ߲��������ϸ������߲������ԭ߲��֢
http://15169.dis.999120.net ��������
���Լ�Ъ��߲���� ѪҺ��
���Լ�Ъ��߲���������Լ�Ъ��Ѫ���ʲ�
http://15170.dis.999120.net ��������
ȱ����ƶѪ ѪҺ��
ȱ����ƶѪ
http://15171.dis.999120.net ��������
���Ժ�ϸ������֢ ѪҺ��
���Ժ�ϸ������֢����ϸ�����ࣻ��ϸ������֢�����Ժ�ϸ������
http://15172.dis.999120.net ��������
�̷��Ժ�ϸ������֢ ѪҺ��
�̷��Ժ�ϸ������֢���̷��Ժ�ϸ������
http://15173.dis.999120.net ��������
����Ժ�ϸ������֢ ѪҺ��
����Ժ�ϸ������֢
http://15174.dis.999120.net ��������
�Ŵ���������ϸ����ƶѪ ѪҺ��
�Ŵ���������ϸ����ƶѪ���Ŵ��Ը����ɺ�ϸ����ƶѪ���Ŵ���������ϸ����ƶѪ
http://15175.dis.999120.net ��������
�̷���������ϸ����ƶѪ ѪҺ��
�̷���������ϸ����ƶѪ���̷���������ϸ��ƶѪ
http://15176.dis.999120.net ��������
����-�����ۺ��� ѪҺ��
����-�����ۺ�����Pearson�ۺ���������-�����ۺ�֢
http://15177.dis.999120.net ��������
Ѫɫ�� ѪҺ��
Ѫɫ������ͭɫ����ɫ���Ը�Ӳ����Ѫɫ�س���֢���Ŵ���Ѫɫ����ԭ���������ɹ���
http://15178.dis.999120.net ��������
�ط��Էκ���Ѫ���س���֢ ѪҺ��
�ط��Էκ���Ѫ���س���֢��Ceelen�����ط��Էκ���Ѫ���س������ط��Էκ���Ѫ���س���֢���ط��Էκ�ɫӲ���ۺ�����ԭ���Էκ���Ѫ���س���֢
http://15179.dis.999120.net ��������
����ϸ����ƶѪ ѪҺ��
����ϸ����ƶѪ������ƶѪ������ϸ����ƶѪ������ƶ������ϸ��ƶѪ
http://15180.dis.999120.net ��������
��������ת������Ѫ֢ ѪҺ��
��������ת������Ѫ֢��������ת������ȱ��֢
http://15181.dis.999120.net ��������
��Ѫ�� ѪҺ��
ά����Cȱ������scurvy����Ѫ����ά����Cȱ��֢��scorbutus��ascorbicaciddeficiency��avitaminosisC����Ѫ֢������Ѫ��ȱ��������Ѫ��ȱ������ά����CӪ��ȱ�ݲ�
http://15182.dis.999120.net ��������
�����ϰ���ƶѪ ѪҺ��
�����ϰ���ƶѪ������
http://15183.dis.999120.net ��������
������ϸ������֢ ѪҺ��
������ϸ������֢��������ϸ������֢��������Ѫ�����֢
http://15184.dis.999120.net ��������
������ϸ��ȱ�� ѪҺ��
������ϸ��ȱ����������ϸ��ȱ��֢��ά-����ϲ�
http://15185.dis.999120.net ��������
������ϸ�������쳣�ۺ��� ѪҺ��
������ϸ�������쳣�ۺ�����Chediak-Higashisyndrome��Chediak-Higashi�ۺ�������ϸ�������쳣�ۺ�������ϸ���쳣ɫ�ؼ����ۺ�������ϸ���쳣ɫ�ؼ����ۺ�֢����-�������ۺϲ�����-�������ۺ�������-���ۺϲ�����-���ۺ�������-ϣ����������-ϣ����������ϸ�������쳣֢��Ⱥ��������ϸ�������쳣��
http://15186.dis.999120.net ��������
�����ϸ���ۺ��� ѪҺ��
�����ϸ���ۺ�����neutrophilparalysis������ϸ���ۺ����������ϸ���ۺ�֢��������ϸ�����
http://15187.dis.999120.net ��������
���������øȱ��֢ ѪҺ��
���������øȱ��֢�����������øȱ��
http://15188.dis.999120.net ��������
��ϸ��������-6-��������øȱ��֢ ѪҺ��
��ϸ��������-6-��������øȱ��֢
http://15189.dis.999120.net ��������
C5���ܲ�ȫ�ۺ��� ѪҺ��
C5���ܲ�ȫ�ۺ�����C5���ܲ�ȫ�ۺ�֢��������C5����ȱ��
http://15190.dis.999120.net ��������
ԭ���Ծ���Ѫ֢ ѪҺ��
ԭ���Ծ���Ѫ֢��Waldenström����Ѫ֢�����Ͼ���Ѫ֢
http://15191.dis.999120.net ��������
��Ӧ�Խ�ϸ������֢ ѪҺ��
��Ӧ�Խ�ϸ������֢����Ӧ�Խ�ϸ������
http://15192.dis.999120.net ��������
��Ѫϸ���ۺ��� ѪҺ��
��Ӧ����֯ϸ������֢����Ӧ����֯ϸ����������Ѫϸ���ۺ�������Ѫ�ۺ���
http://15193.dis.999120.net ��������
ԭ���Ե���¡�����ײ� ѪҺ��
ԭ���Ե���¡�����ײ���benignmonoclonalimmunoglobulinopathy��MIUS��monoclonalimmunoglobulinopathyofunknownsignificance�����Ե���¡�����ײ������岻���Ե���¡�����ײ���ԭ���Ե���¡�����ײ���ԭ���Ե���¡������Ѫ֢��ԭ�����ĵ���¡����������֢
http://15194.dis.999120.net ��������
�̷��Ե���¡�����ײ� ѪҺ��
�̷��Ե���¡�����ײ����鷢�ڷǽ�ϸ���Լ����ĵ���¡������Ѫ֢�̷��Ե���¡���ײ����̷��Ե���¡������Ѫ֢
http://15195.dis.999120.net ��������
�������� ѪҺ��
���������ԣ�amyloiddegeneration���������䣻���������Բ���amyloidthesaurismosis��baconydegeneration��cellulosedegeneration��chitinousdegeneration��gammaloidosis��glassyswelling��hyaloiddegeneration��lardaceousdegeneration��waxydegeneration��������������������
http://15196.dis.999120.net ��������
Ѫ������ĸϸ�����ܰͽᲡ ѪҺ��
Ѫ������ĸϸ�����ܰͽᲡ��AILDwithdysproteinmia��lymphogranulomatosisX���ܰ���ѿ��X��Ѫ������ĸϸ�����ܰͽᲡ���쳣����Ѫ֢
http://15197.dis.999120.net ��������
Castleman�� ѪҺ��
Castleman����vascularfollicularlymphnodehyperplasia�����ܰͽ�������Ѫ���������ܰͽ�������
http://15198.dis.999120.net ��������
������֯ϸ���� ѪҺ��
������֯ϸ������Tϸ���ܰ�����������״ϸ������������֯ϸ������֢�����飻�ǰ�Ѫ����״��Ƥϸ������֢���ܰ���״����������״ϸ����Ѫ������֯ϸ����������״ϸ������֢
http://15199.dis.999120.net ��������
�ʸ�˹��֯ϸ������֢ ѪҺ��
�ʸ�˹��֯ϸ������֢��Letterer-Siwedisease��LSD����-ѩ������֯ϸ������֢X
http://15200.dis.999120.net ��������
������ϸ������֢ ѪҺ��
������ϸ������֢������ϸ������֢
http://15201.dis.999120.net ��������
�ȼ�����ϸ������֢ ѪҺ��
�ȼ�����ϸ������֢���ȼ�����ϸ������
http://15202.dis.999120.net ��������
������ϸ������֢ ѪҺ��
������ϸ������֢��������ϸ����
http://15203.dis.999120.net ��������
�ط�����������ϸ�������ۺ��� ѪҺ��
�ط�����������ϸ�������ۺ������ط�����������ϸ�������ۺ�֢����������ϸ���ۺ������ط���������ϸ�������ۺ���
http://15204.dis.999120.net ��������
����������ϸ������֢ ѪҺ��
����������ϸ������֢��eosinophiliclungdisease���ΰ�������ϸ������ν�������ϸ���ԷΣ���������ϸ���Էμ���
http://15205.dis.999120.net ��������
������������ϸ���Է��� ѪҺ��
������������ϸ���Է��ף�����������ϸ������
http://15206.dis.999120.net ��������
������������ϸ���Է��� ѪҺ��
������������ϸ���Է��ף�prolongedeosinophiliapneumonia��������ϸ���Է��ף����������Է��ף�Ǩ����������ϸ������֢��Ǩ���Է���������ϸ������֢
http://15207.dis.999120.net ��������
�����Է���������ϸ������֢ ѪҺ��
�����Է���������ϸ������֢��Löffler��ssyndrome��Löffler�ۺ���
http://15208.dis.999120.net ��������
�ȴ��Է���������ϸ������֢ ѪҺ��
�ȴ��Է���������ϸ������֢��Weingarten�ۺ������ȴ�������ϸ�����ࣻ�ȴ������ϸ������֢
http://15209.dis.999120.net ��������
����ϸ����θ���� ѪҺ��
����ϸ����θ���ף�������ϸ����θ���ף���������ϸ����θ����
http://15210.dis.999120.net ��������
ԭ���Թ�����ά�� ѪҺ��
ԭ���Թ�����ά��
http://15211.dis.999120.net ��������
�̷��Թ�����ά�� ѪҺ��
�̷��Թ�����ά��
http://15212.dis.999120.net ��������
���̲� ѪҺ��
���������ȱ���ۺ�����AIDS�����̲������������ȱ���ۺ�֢����������߹���ɥʧ֢��HIV��Ⱦ
http://15213.dis.999120.net ��������
Ƣ���ܿ��� ѪҺ��
Ƣ���ܿ�����Ƣ���ܿ���
http://15214.dis.999120.net ��������
��л�� ѪҺ��
��л����������Ƣ��ƶѪ������������֬��������������֬øȱ��֢��cerebrosidelipoidosis��cerebrosidosis��familialsplenicanemia��Gaucher����glucosylceramidelipoidosis����лƢ�״���߰������߰������������֬������������֬����֢������֬��״��Ƥϸ��������������������
http://15215.dis.999120.net ��������
����-Ƥ�˲� ѪҺ��
����-Ƥ�˲���Niemann-Pick����sphingomyelinstoragedisease����֬��֯ϸ������֢������-Ƥ�˶��ϲ�����-Ƥ������-Ƥ���ϲ�������֬���˾���ϸ����������֬��������������֬��������������֬���۲���sphingomyelinosis����������֬��л�ϰ���niemann-pick��sdisease������֬
http://15216.dis.999120.net ��������
��ֲ���ܰ���ֳ�Լ��� ѪҺ��
��ֲ���ܰ���ֳ�Լ�������ֲ���ܰ��������ϰ�
http://15217.dis.999120.net ��������
�����֯ϸ����������ܰͽᲡ ѪҺ��
�����֯ϸ����������ܰͽᲡ��Rosai-Dorfman�ۺ��������֯ϸ������֢���д�����ܰͽᲡ
http://15219.dis.999120.net ��������
ԭ�������Լ�����ص��ܰ���ֳ�Լ��� ѪҺ��
ԭ�������Լ�����ص��ܰ���ֳ�Լ���
http://15220.dis.999120.net ��������
��������E�ۺ��� ѪҺ��
��������E�ۺ�����Buckley�ۺ�����granulomatousdiseasevariant��hyperimmunoglobulinemiaEsyndrome��Job�ۺ�������IgE�ۺ�������������EѪ֢�ۺ�������������E�ۺ�֢��������ѿ�ײ������ͣ�ҦƤ���ۺ���
http://15221.dis.999120.net ��������
��Ⱦ���ܰ�ϸ������֢ ѪҺ��
��Ⱦ���ܰ�ϸ������֢
http://15222.dis.999120.net ��������
������ѿ�ײ� ѪҺ��
������ѿ�ײ�����ͯ������ѿ�ף�������ѿ���Լ���
http://15223.dis.999120.net ��������
�����Խ�ڲ�����ѿ�ײ� ѪҺ��
�����Խ�ڲ�����ѿ�ײ�
http://15224.dis.999120.net ��������
������ ѪҺ��
��������heavy-chaindisease��Fc���Franklin����Seligmann��������������������������������
http://15225.dis.999120.net ��������
����Ѫ�� ѪҺ��
�����ܰ�ϸ����Ѫ�������ܣ����Գ��ܰ�ϸ����Ѫ����ALL������Ѫ��
http://15226.dis.999120.net ��������
������ܰ�ϸ����Ѫ�� ѪҺ��
������ܰ�ϸ����Ѫ����T���ܰ�ϸ����ֳ�Լ�����T�����ܰ�ϸ����Ѫ����������ܰ�ϸ�����ܰ�ϸ����ֳ�Լ�����������ܰ�ϸ����Ѫ��
http://15227.dis.999120.net ��������
�ʴ�ϸ����Ѫ�� ѪҺ��
�ʴ�ϸ����Ѫ������֯�ȼ�ϸ����Ѫ��
http://15228.dis.999120.net ��������
��ϸ����Ѫ�� ѪҺ��
��ϸ����Ѫ������ϸ����Ѫ����Plasmacyticleukemia��PCL
http://15229.dis.999120.net ��������
��������ϸ����Ѫ�� ѪҺ��
��������ϸ����Ѫ��������ϸ����Ѫ��
http://15230.dis.999120.net ��������
�ȼ�����ϸ����Ѫ�� ѪҺ��
�ȼ�����ϸ����Ѫ�����ȼ�ϸ����Ѫ����basophiliccellleukemia
http://15231.dis.999120.net ��������
������Ѫ�� ѪҺ��
������Ѫ����congenitalleukemi��CL
http://15232.dis.999120.net ��������
�� ѪҺ��
������ϵͳ��Ѫ������
http://15233.dis.999120.net ��������
�̷���Ѫ�� ѪҺ��
�̷���Ѫ����t-MDS/AML��������ع��������쳣�ۺ���/������ϵ��Ѫ��
http://15234.dis.999120.net ��������
�ܰ���ϸ����Ѫ�� ѪҺ��
�ܰ���ϸ����Ѫ����LSL��lymphosarcomacellleukemia���ܰ�����ϸ����Ѫ��
http://15235.dis.999120.net ��������
����Tϸ����Ѫ�� ѪҺ��
����Tϸ����Ѫ��������Tϸ����Ѫ����������Tϸ����Ѫ����adultT-cellleukemia/lymphoma������Tϸ����Ѫ��/�ܰ���
http://15236.dis.999120.net ��������
������ϸ����Ѫ�� ѪҺ��
������ϸ����Ѫ����ANLL�������ܣ����������Է��ܰ�ϸ����Ѫ����������ϸ����Ѫ��
http://15237.dis.999120.net ��������
�������Լ���Ѫ�� ѪҺ��
�������Լ���Ѫ������Ѫ��ǰ�ڣ�ð����Ѫ����HAL
http://15238.dis.999120.net ��������
���Ѫ����Ӧ ѪҺ��
���Ѫ����Ӧ����Ѫ������Ӧ
http://15239.dis.999120.net ��������
�����Ӻ���Ѫ�� ѪҺ��
�����Ӻ���Ѫ����acutemixedleukemia�����Ի��ϸ����Ѫ�����ӽ��Լ���Ѫ��
http://15240.dis.999120.net ��������
���꼱��Ѫ�� ѪҺ��
���꼱��Ѫ����senileacuteleucemia
http://15241.dis.999120.net ��������
����������ϸ����Ѫ�� ѪҺ��
����������ϸ����Ѫ��������ǰ��ϸ����Ѫ��������������ϸ����Ѫ����APL
http://15242.dis.999120.net ��������
���ܰ�ϸ����Ѫ�� ѪҺ��
���ܰ�ϸ����Ѫ����ǰ�ܰ�ϸ����Ѫ�������ܰ�ϸ����Ѫ��
http://15243.dis.999120.net ��������
ëϸ����Ѫ�� ѪҺ��
ëϸ����Ѫ����ëϸ����Ѫ������Ѫ������״��Ƥϸ����������ëϸ����Ѫ����hairy-cellleukemia
http://15244.dis.999120.net ��������
������ϸ����Ѫ�� ѪҺ��
������ϸ����Ѫ����������������ϸ����Ѫ����������ϸ����Ѫ����������ϸ����Ѫ��
http://15245.dis.999120.net ��������
�����ܰ�ϸ����Ѫ�� ѪҺ��
�����ܰ�ϸ����Ѫ�������ܣ������ܰ�ϸ����Ѫ����CLL
http://15246.dis.999120.net ��������
�����ܰ��� ѪҺ��
�����ܰ���
http://15247.dis.999120.net ��������
����� ѪҺ��
������νܽ�������ܰͲ����ܰ���ѿ�ף��ܰ���״ϸ��������-˹����Hodgkin'sdisease���νܽ��ϲ�
http://15248.dis.999120.net ��������
ԭ����Ƥ��Bϸ���ܰ��� ѪҺ��
ԭ����Ƥ��Bϸ���ܰ�����primarycutaneousB-celllymphoma
http://15249.dis.999120.net ��������
ަ���������Sezary�ۺ��� ѪҺ��
ަ���������Sezary�ۺ�����ަ��ù������Sezary�ۺ�����ަ����ѿ�ײ���Sezary�ۺ�����ަ���������Sezary�ۺ�֢
http://15250.dis.999120.net ��������
ԭ�����������ܰ��� ѪҺ��
ԭ�����������ܰ�����lymphomaprimaryeffusion��primaryeffusionlymphoma
http://15251.dis.999120.net ��������
Ƥ��֬Ĥ����Tϸ���ܰ��� ѪҺ��
Ƥ��֬Ĥ����Tϸ���ܰ�����Ƥ��֬Ĥ����T-ϸ���ܰ�����subcutaneouspanniculitictcelllymphoma
http://15252.dis.999120.net ��������
���������ȱ���ۺ�������ܰ��� ѪҺ��
���������ȱ���ۺ�������ܰ��������̲�����ܰ��������������ȱ���ۺ�������ܰ���
http://15253.dis.999120.net ��������
ԭ���Թ��ܰ��� ѪҺ��
ԭ���Թ��ܰ�����ԭ���Թ���״ϸ��������primarylymphomaofbone
http://15255.dis.999120.net ��������
�ܰ�������ѿ�� ѪҺ��
�ܰ�������ѿ�ף��ܰ�������ѿ�ײ���polymorphicreticulosis����������״ϸ������֢������Ѫ������ѿ�ײ���Ѫ���������ܰ�����Ѫ��������������ֳ�Բ��䣻������״ϸ������
http://15256.dis.999120.net ��������
�μ����ܰ��� ѪҺ��
�μ����ܰ�����nodularlymphoidhyperplasia�����ܰ�����������ܰ���֯���������μ��ܰ�����������ܰ���֯����
http://15257.dis.999120.net ��������
�ǻ�����ܰ��� ѪҺ��
�ǻ�����ܰ������Ǻνܽ��ܰ������ǻ��������ܰ�������NHL
http://15258.dis.999120.net ��������
�Ĥ�������֯�ܰ��� ѪҺ��
�Ĥ�������֯�ܰ�����ճĤ�������֯�ܰ������Ĥ�������֯�ܰ�������ճĤ�������֯�ܰ�����
http://15259.dis.999120.net ��������
ԭ���������ܰ��� ѪҺ��
ԭ���������ܰ���
http://15260.dis.999120.net ��������
ԭ����������ϵͳ�ܰ��� ѪҺ��
ԭ����������ϵͳ�ܰ�������״ϸ��������С����ϸ������Ѫ��������
http://15261.dis.999120.net ��������
غ���ܰ��� ѪҺ��
غ���ܰ�����testiclymphadenoma��غ���ܰ�����
http://15262.dis.999120.net ��������
ԭ���Ա�ǻ�ܰ��� ѪҺ��
ԭ���Ա�ǻ�ܰ���
http://15263.dis.999120.net ��������
ԭ���������ܰ��� ѪҺ��
ԭ���������ܰ���
http://15265.dis.999120.net ��������
�������ܰͽ��� ѪҺ��
��֯ϸ���������ܰͽ��ף�Kikuchi-Fujimoto����Kikuchi�����������ܰͽ��ף��������ܰͽ��ף��Ǽ����ܰͽ���
http://15266.dis.999120.net ��������
��Ⱦ�Ե���ϸ������֢ ѪҺ��
��Ⱦ�Ե���ϸ������֢����Ⱦ�Ե���ϸ�����ࣻacutebenignlymphoblastosis��acutelymphadenosis��febrisglandularis��lymphaticreaction��mononucleosisinfectiosa��Pfeiffer'sdisease��Turck'ssyndrome�����������ϲ����������Գ��ܰ�ϸ������֢�������ܰ���֯�������ؿ��ۺ���
http://15267.dis.999120.net ��������
��Թ����� ѪҺ��
��Թ�������Kahler�����������������ز����������ϲ�����ϸ��������
http://15268.dis.999120.net ��������
��������� ѪҺ��
��������anaphylacticpurpura��Henoch-Schonleinsyndrome����ŵ-�����ۺ���������Ѫ������anaphylactoidpurpura��Henoch-Schonlein����̬��Ӧ������Ӧ���������ߣ�Ѫ�累��Henoch-Schonlein�ۺ�������Ѫ��ëϸѪ���ж�֢
http://15269.dis.999120.net ��������
�����յ���ѪС�����֢ ѪҺ��
�����յ���ѪС�����֢�������շ���ѪС�����֢
http://15270.dis.999120.net ��������
����״Ѫ����ѪС������ۺ��� ѪҺ��
����״Ѫ����ѪС������ۺ���������״Ѫ����ѪС������ۺ�֢
http://15271.dis.999120.net ��������
ѪС����������쳣 ѪҺ��
ѪС����������쳣��Scott�ۺ�����ѪС���3����ȱ��֢��ѪС��ڢ�����ȱ��֢
http://15272.dis.999120.net ��������
Ѫ˨��ѪС���������� ѪҺ��
Ѫ˨��ѪС���������Ѫ˨�γ���ѪС����������
http://15273.dis.999120.net ��������
ѪС������֢ ѪҺ��
ѪС������֢��Glanzmannthrombasthenia��Glanzmann����GT��ѪС�幦�ܲ�ȫ��ѪС����ܲ�ȫ
http://15274.dis.999120.net ��������
��ѪС�岡 ѪҺ��
��ѪС�岡��Bernard-Soulier�ۺ���
http://15275.dis.999120.net ��������
��-����ز� ѪҺ��
��-����ز�����-����ز�����-����ⲡ����ɫѪС���ۺ���
http://15276.dis.999120.net ��������
��-����ز� ѪҺ��
��-����ز�����-����ز�����-����ⲡ
http://15277.dis.999120.net ��������
Ѫ�Ѳ�A ѪҺ��
Ѫ�Ѳ��ף�Ѫ�Ѳ�A
http://15278.dis.999120.net ��������
Ѫ�Ѳ��� ѪҺ��
Ѫ�Ѳ��ң�Christmas����Ѫ����Ѫ��ø�ɷ�ȱ��֢��Ѫ�Ѳ�B���Ŵ������Ӣ�ȱ��֢
http://15279.dis.999120.net ��������
�����Ѫ�Ѳ� ѪҺ��
�����Ѫ�Ѳ�
http://15280.dis.999120.net ��������
ѪС���ͼ���Ѫ����Ѫ�Ѳ� ѪҺ��
ѪС���ͼ���Ѫ����Ѫ�Ѳ�������Ѫ����Ѫ�Ѳ���ѪС����Ѫ����Ѫ�Ѳ�
http://15281.dis.999120.net ��������
��������Ѫ֢ ѪҺ��
��������Ѫ֢��hemorrhagicdiseaseofnewborn����������Ѫ��������������ѪøԭѪ֢���������ڱ㣻��������Ȼ��Ѫ֢
http://15282.dis.999120.net ��������
�Ŵ�����Ѫøԭȱ�� ѪҺ��
�Ŵ�����Ѫøԭȱ�����Ŵ�����Ѫ���Ӣ�ȱ��
http://15283.dis.999120.net ��������
�Ŵ�����Ѫ���Ӣ�ȱ��֢ ѪҺ��
�Ŵ�����Ѫ���Ӣ�ȱ��֢���Ŵ�����Ѫ���Ӣ�ȱ��
http://15284.dis.999120.net ��������
�Ŵ�����Ѫ���Ӣ�ȱ�� ѪҺ��
�Ŵ�����Ѫ���Ӣ�ȱ��
http://15285.dis.999120.net ��������
�Ŵ�����Ѫ���Ӣ�ȱ�� ѪҺ��
�Ŵ�����Ѫ���Ӣ�ȱ��
http://15286.dis.999120.net ��������
�Ŵ�����Ѫ���Ӣ�ȱ�� ѪҺ��
�Ŵ�����Ѫ���Ӣ�ȱ����Ѫ�Ѳ�C
http://15287.dis.999120.net ��������
�Ŵ�����Ѫ���Ӣ���ȱ�� ѪҺ��
�Ŵ�����Ѫ���Ӣ���ȱ��
http://15288.dis.999120.net ��������
�Ŵ��Գ�Ѫ��ëϸѪ������ ѪҺ��
�Ŵ��Գ�Ѫ��ëϸѪ�����ţ�Rendu-Osler-Weber����telangiectasiahereditariahaemorrhagica���Ŵ��Գ�Ѫ��ëϸѪ������֢��Osler-Rendu-Weber�ۺ������Ŵ���ëϸѪ������֢
http://15289.dis.999120.net ��������
�Ŵ�����ά����ԭȱ��֢ ѪҺ��
�Ŵ�����ά����ԭȱ��֢���Ŵ�����ά����ԭ���Ӣ�ȱ��֢���Ŵ������Ӣ�ȱ��֢
http://15290.dis.999120.net ��������
�Ŵ����쳣��ά����ԭѪ֢ ѪҺ��
�Ŵ����쳣��ά����ԭѪ֢
http://15291.dis.999120.net ��������
�Ŵ��Կ���Ѫø��ȱ��֢ ѪҺ��
�Ŵ��Կ���Ѫø��ȱ��֢
http://15292.dis.999120.net ��������
�Ŵ��Ե���Cȱ��֢ ѪҺ��
�Ŵ��Ե���Cȱ��֢���Ŵ��Ե���Cȱ��֢
http://15293.dis.999120.net ��������
�Ŵ��Ե���Sȱ��֢ ѪҺ��
�Ŵ��Ե���Sȱ��֢
http://15294.dis.999120.net ��������
����ĵ���C֢ ѪҺ��
����ĵ���C֢��APC�ֿ�
http://15295.dis.999120.net ��������
�����ά����K��������Ѫ�����쳣 ѪҺ��
�����ά����K��������Ѫ�����쳣
http://15296.dis.999120.net ��������
�����ѭ��������������֢ ѪҺ��
�����ѭ��������������֢
http://15297.dis.999120.net ��������
���ظβ�����������Ѫ�����쳣 ѪҺ��
���ظβ�����������Ѫ�����쳣
http://15298.dis.999120.net ��������
ԭ������ά�����ܽ�֢ ѪҺ��
ԭ������ά�����ܽ�֢��ԭ��������
http://15299.dis.999120.net ��������
������Ѫ������Ѫ ѪҺ��
��ɢ��Ѫ������Ѫ��consumptioncoagulopathy����ɢ��Ѫ������Ѫ��ȥ��ά����ԭ�ۺ�������������Ѫ����defibrinationsyndrome��diffuseintravascularcoagulation��������Ѫ������Ѫ��ȥ��ά�����ۺ�����DIC
http://15300.dis.999120.net ��������
ԭ����ѪС������֢ ѪҺ��
ԭ����ѪС������֢
http://15301.dis.999120.net ��������
Ѫ˨�γ� ѪҺ��
Ѫ˨�γɣ�Ѫ˨֢
http://15303.dis.999120.net ��������
�ط���ѪС���������� ѪҺ��
�ط���ѪС���������immunethrombocytopenicpurpura��������ѪС���������ԭ����ѪС����������
http://15304.dis.999120.net ��������
��Ⱦ��ѪС���������� ѪҺ��
��Ⱦ��ѪС����������
http://15305.dis.999120.net ��������
��������� ѪҺ��
���������
http://15306.dis.999120.net ��������
��Ѫ��ѪС���������� ѪҺ��
��Ѫ��ѪС���������post-transfusionalpurpura����Ѫ�����
http://15307.dis.999120.net ��������
������ϸ����������� ѪҺ��
������ϸ����������Gardner-Diamond�ۺ���������������ʹ�Է����������ٰ�����������ϸ����̬��Ӧ�����
http://15308.dis.999120.net ��������
ҩ��������ѪС���������� ѪҺ��
ҩ��������ѪС����������
http://15309.dis.999120.net ��������
ͬ��������������ѪС���������� ѪҺ��
ͬ��������������ѪС���������������ͬ������ѪС����������
http://15310.dis.999120.net ��������
����ճ�� �ۿ�
����ճ��
http://15311.dis.999120.net ��������
�����ٰ� �ۿ�
����Ƥ֬�ٰ��������ٰ��������ٰ���Sebaceouscarcinomaofeyelid
http://15312.dis.999120.net ��������
��������ϸ���� �ۿ�
��������ϸ������Gorlin-Goltz�ۺ���
http://15313.dis.999120.net ��������
�������Ժ�ɫ��ϸ������ �ۿ�
�������Ժ�ɫ��ϸ���������������Ժ�ɫ����
http://15314.dis.999120.net ��������
����Ĥ�������� �ۿ�
����Ĥ��������������Ĥ�������
http://15315.dis.999120.net ��������
����Ĥ�������� �ۿ�
����Ĥ��������������Ĥ��������
http://15316.dis.999120.net ��������
���������Ĥ������ �ۿ�
���������Ĥ������
http://15317.dis.999120.net ��������
coats�� �ۿ�
�������������Ĥ���䣻coats����externalhemorrhagicretinopathy��retinaltelangiectasis������ĤëϸѪ������֢������Ѫ������Ĥ����
http://15318.dis.999120.net ��������
�����������Բ���������Ĥ���� �ۿ�
�����������Բ���������Ĥ����
http://15319.dis.999120.net ��������
���������Ĥ���� �ۿ�
���������Ĥ���䣻��״�����ά����֢�����������Ĥ��
http://15320.dis.999120.net ��������
����������Ĥ���� �ۿ�
����������Ĥ���䣻����������Ĥ��
http://15321.dis.999120.net ��������
�ư�����ˮ�� �ۿ�
�ư�����ˮ�ף������ư�ˮ��
http://15322.dis.999120.net ��������
�����Իư߱��� �ۿ�
�����Իư߱��ԣ�senilemaculardegeneration�������Իư߱��ԣ���������Իư߱���
http://15323.dis.999120.net ��������
�����Խ�Һ������Ĥ����Ĥ���� �ۿ�
�����Խ�Һ������Ĥ����Ĥ���䣻����������������Ĥ�ף��ط��������Խ�Һ������Ĥ�ף��н�
http://15324.dis.999120.net ��������
����������������Ĥ����Ĥ���� �ۿ�
����������������Ĥ����Ĥ���䣻Rieger������������������Ĥ����Ĥ�ף�����
http://15325.dis.999120.net ��������
����Ĥ������Ѫ��Ĥ �ۿ�
����Ĥ������Ѫ��Ĥ��choroidalneovascularization��CNV������Ĥ����Ѫ��Ĥ�γ�
http://15326.dis.999120.net ��������
�ط��Իư��ѿ� �ۿ�
�ط��Իư��ѿ�
http://15327.dis.999120.net ��������
�ѻ�״�ư�Ӫ������ �ۿ�
�ѻ�״�ư�Ӫ��������Best����polymorphicmaculardegeneration�����λư߱���
http://15328.dis.999120.net ��������
Stargardt�� �ۿ�
Stargardt����fundusflavimaculatuswithmaculardystrophy����ɫ�ߵ�״�۵ϲ��ư߱��ԣ�˹�����ز���˹�������ϲ����۵�ɫ�ߵ�֢�����Իư�Ӫ�������������Ŵ��Իư�Ӫ������
http://15329.dis.999120.net ��������
�ط��Իư߲�ǰĤ �ۿ�
�ط��Իư߲�ǰĤ��idioputhicpreretinalmacularfibrosis��preretinalmembrane��primaryretinalfolds������ĤǰĤ���ط��Իư�ǰĤ���ط��Իư�����ĤǰĤ���ط�������Ĥǰ�ư߲���ά����ԭ��������Ĥ����
http://15330.dis.999120.net ��������
����Ĥɫ�ر��� �ۿ�
ԭ��������Ĥɫ�ر��ԣ�retinitispigmentosa��ɫ��������Ĥ�ף�����Ĥɫ�ر���
http://15331.dis.999120.net ��������
���������Ĥ���� �ۿ�
���������Ĥ���ѣ�senileretinoschisis������������Ĥ����֢
http://15332.dis.999120.net ��������
����������Ĥ����֢ �ۿ�
����������Ĥ����֢��congenitalvascularveilsinthevitreum��hereditaryretinoschisis��juvenileretinoschisis������������Ĥ����֢�������Բ�����Ѫ��ɴĤ֢���Ŵ�������Ĥ����
http://15333.dis.999120.net ��������
�����Խ��ӵ��۵��� �ۿ�
�����Խ��ӵ��۵��������Խ��������۵��������Խ��ӵ��۵���
http://15334.dis.999120.net ��������
��Ѫѹ������Ĥ���� �ۿ�
��Ѫѹ������Ĥ���䣻��Ѫѹ����Ĥ����
http://15335.dis.999120.net ��������
���Ը�Ѫѹ����Ĥ���� �ۿ�
���Ը�Ѫѹ����Ĥ���䣻retinopathyduetoacceleratedhypertension��������Ѫѹ����Ĥ����
http://15336.dis.999120.net ��������
�����Ѫѹ�ۺ����۵ײ��� �ۿ�
�����Ѫѹ�ۺ����۵ײ��䣻retinopathyduetopregnancy-inducedhypertensionsyndrome��retinopathyduetotoxeniaofpregnancy���Ѹ�������Ĥ���䣻�����ж�֢����Ĥ����
http://15337.dis.999120.net ��������
������������խ����Ĥ���� �ۿ�
������������խ����Ĥ���䣻retinopathyduetohypoperfusionretinopathy����ע����Ĥ��������Ĥ����
http://15338.dis.999120.net ��������
���������ۺ�������Ĥ���� �ۿ�
���������ۺ�������Ĥ���䣻Martorell�ۺ�������Ĥ���䣻retinopathyduetopulselessdisease��retinopathyduetoTakayasu'sdisease��Takayasu������Ĥ���䣻��Դ���������Ĥ���䣻����֢����Ĥ����
http://15339.dis.999120.net ��������
����������Ĥ���� �ۿ�
����������Ĥ���䣻����������Ĥ��
http://15340.dis.999120.net ��������
��ϸ������֢����Ĥ���� �ۿ�
��ϸ������֢����Ĥ���䣻Vaquez-Osler������Ĥ���䣻��ϸ����������Ĥ����
http://15341.dis.999120.net ��������
��Ѫ������Ĥ���� �ۿ�
��Ѫ������Ĥ���䣻retinopathyduetoleucosis����ϸ����֯����֢����Ĥ���䣻Ѫ������Ĥ����
http://15342.dis.999120.net ��������
��״ϸ��ƶѪ�۲����� �ۿ�
��״ϸ��ƶѪ�۲�����
http://15343.dis.999120.net ��������
���������ȱ���ۺ�������Ĥ���� �ۿ�
���������ȱ���ۺ�������Ĥ���䣻���̲�����Ĥ����
http://15344.dis.999120.net ��������
����Ĥ���� �ۿ�
����Ĥ���룻����Ĥ���룻����Ĥ����Ƥ������
http://15345.dis.999120.net ��������
�ư��ѿ�������Ĥ���� �ۿ�
�ư��ѿ�������Ĥ����
http://15346.dis.999120.net ��������
���ѿ�������Ĥ���� �ۿ�
���ѿ�������Ĥ����
http://15347.dis.999120.net ��������
�����Բ���������Ĥ���� �ۿ�
�����Բ���������Ĥ���䣻massiveperiretinalproliferation��massivepreretinalretraction��massivevitreousretraction���㷺������ǣ�����㷺�������������㷺����Ĥǰǣ�����㷺����Ĥ��Χ��ֳ���㷺������Ĥǰ�������㷺������Ĥ��Χ��������ֳ�Բ���������Ĥ����
http://15349.dis.999120.net ��������
����Ĥ��Ĥ �ۿ�
����Ĥ��Ĥ������Ĥ��Ĥ
http://15350.dis.999120.net ��������
����Ĥĸϸ���� �ۿ�
����Ĥĸϸ������������Ĥϸ����������Ĥ����ϸ������retinalglioblastoma��retinalneuroblastoma��retinoma������Ĥ���Խ�����
http://15351.dis.999120.net ��������
����ĤëϸѪ��Ѫ���� �ۿ�
����ĤëϸѪ��Ѫ������vonHippel��������Ĥ��ɫ��
http://15352.dis.999120.net ��������
�������Ѫ �ۿ�
�������Ѫ���������Ѫ
http://15353.dis.999120.net ��������
Terson�ۺ��� �ۿ�
Terson�ۺ�����Terson�ۺ�֢
http://15354.dis.999120.net ��������
Զ�� �ۿ�
Զ�ӣ�farsight��farsightedness��longsight��longsightedness��long-sightedness��DZ����Զ�ӣ�����Զ��
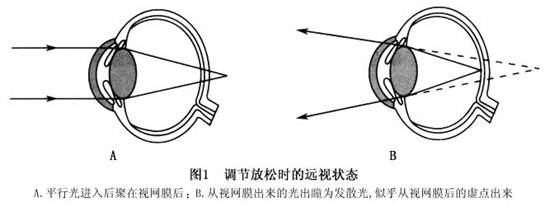
http://15355.dis.999120.net ��������
���� �ۿ�
���ӣ����ӣ������ۣ�������
http://15356.dis.999120.net ��������
ɢ�� �ۿ�
ɢ�⣻������
http://15357.dis.999120.net ��������
����β� �ۿ�
����β����β�֢
http://15358.dis.999120.net ��������
��״���� �ۿ�
��״����
http://15359.dis.999120.net ��������
�ϻ��� �ۿ�
���ӣ��ϻ���
http://15360.dis.999120.net ��������
��б�� �ۿ�
��б��latentsquint��phoria��recessivedeflection��suppressedstrabismus��DZ����б�ӣ�������б�ӣ���ƫ���б��
http://15361.dis.999120.net ��������
��ͬ��б�� �ۿ�
��ͬ��б��
http://15362.dis.999120.net ��������
�����б�� �ۿ�
�����б�ӣ�paralyticsquint
http://15363.dis.999120.net ��������
��������ۺ��� �ۿ�
��������ۺ�����Duaneretractionsyndrome��Duane��������ۺ�����retractionstrabismus��������б�ӣ�ʩ�������ۺ�����ʩ�����ۺ�������������˶��ۺ�������������ۺ�֢�����������ۺ���
http://15364.dis.999120.net ��������
��б�������ۺ��� �ۿ�
��б�������ۺ�����Brownsuperiortendonsheathsyndrome��Brownsyndrome��Brown�ۺ������������ۺ����������ۺ�������б�������ۺ�֢����б��������Ⱥ
http://15365.dis.999120.net ��������
�̶���б�� �ۿ�
�̶���б�ӣ��̶�б��
http://15366.dis.999120.net ��������
���⼡�㷺��ά���ۺ��� �ۿ�
���⼡�㷺��ά���ۺ�����alldysplasiaofocularmuscl��congenitalgeneralfibrosisofextraocularmusclessyndrome���㷺��ά���ۺ�����ȫ���⼡������ȫ���������۲���ά���ۺ��������������⼡�㷺��ά���ۺ��������������⼡��ȫ��ά���ۺ��������⼡�㷺��ά���ۺ�֢�����⼡�ձ���ά����
http://15367.dis.999120.net ��������
�������ۿ����� �ۿ�
�������ۿ����ۣ�orbitalfloorfracturesyndrome�������Կ����ۣ����ױ��ۣ��������ۺ���
http://15368.dis.999120.net ��������
�ۿ���֢������ �ۿ�
�ۿ���֢��������myasthenicpseudoparalysisofophthalmology���ۿƼ��������֢���������ۿ���֢������֢
http://15369.dis.999120.net ��������
���Խ��������⼡��� �ۿ�
���Խ��������⼡��ԣ�vonGraefe�ۼ��������Խ��������ۼ���ԣ����Խ��������⼡���֢�����⼡������Ӫ��������
http://15370.dis.999120.net ��������
��������б�� �ۿ�
��������б�ӣ�alternate-dayesotropia��clock-mechanismesotropiac��cyclicesoropia��ircadianesotropia����������б�ӣ�������б�ӣ�������������б�ӣ�ʱ�ӻ�������б�ӣ�������б��
http://15371.dis.999120.net ��������
���Թ�ͬ��б�� �ۿ�
���Թ�ͬ��б��
http://15372.dis.999120.net ��������
A-V�ۺ��� �ۿ�
A-V�ۺ�����AandVphenomenon��AandVsign��AandVstrabismus��A-V����A-V��б�ӣ�A-V����AV�ۺ�����A-V�ۺ�֢��Uristsyndrome��Urist�ۺ���
http://15373.dis.999120.net ��������
�����Դ�ֱƫб �ۿ�
�����Դ�ֱƫб�����봹ֱ��ƫб
http://15374.dis.999120.net ��������
����б�� �ۿ�
�����
http://15375.dis.999120.net ��������
С����б�� �ۿ�
С����б�ӣ�microstrabismus��monofixationsyndrome��parkssy��Parks֢��Ⱥ��smallangle��С�Ƕȣ�ultrasmallanglestrabismus����С�Ƕ�б�ӣ�����̶����ۺ���������ע���ۺ������̶����죻С�Ƕ�б�ӣ�б������ע���ۺ�����б��
http://15376.dis.999120.net ��������
Ӥ������б�� �ۿ�
Ӥ������б�ӣ�congenitalesotropia����������б�ӣ�������Ӥ������б�ӣ�Ӥ������б���ۺ���
http://15377.dis.999120.net ��������
������������ۺ��� �ۿ�
������������ۺ�����������������ۺ�֢
http://15378.dis.999120.net ��������
�����Զ�������� �ۿ�
�����Զ�������ԣ�Axenfeld-Schurenberg�ۺ�����cyclicoculomotorspasmrelaxationphenomenon��cyclicoculomotordisease����-������ۺ����������������������˶���ԣ����ھ����Զ�������ԣ������Զ������������Զ������Σ������Զ������γڻ����������������˶���Լ�
http://15381.dis.999120.net ��������
��ʹ���ۼ���� �ۿ�
��ʹ���ۼ���ԣ�Tolosa-Hunt�ۺ�����������ף���ʹ���ۼ�����ۺ����������ۼ���ԣ���-������ۺ���
http://15383.dis.999120.net ��������
˫����б����� �ۿ�
˫����б�����
http://15384.dis.999120.net ��������
�ۿ����� �ۿ�
�ۿ����ף�externalmyositis�����⼡��
http://15385.dis.999120.net ��������
���⼡������ȫ �ۿ�
���⼡������ȫ��ateloabsentofocularmuscles��dysplasiaofocularmuscles�����⼡�������������⼡����ȱ��
http://15386.dis.999120.net ��������
���⼡���� �ۿ�
���⼡���ˣ�externalmyoplegia��traumatic��traumaticparalyticstrabismus��traumaticstrabismus�������������б�ӣ�������б�ӣ����������⼡��ԣ����⼡������
http://15387.dis.999120.net ��������
���⼡����β�ʲ� �ۿ�
���⼡����β�ʲ������⼡�ҳ没�������ҳ没
http://15388.dis.999120.net ��������
���� �ۿ�
���ӣ�dimsightedness��lazyeyes��visushebetudo��weaksight
http://15389.dis.999120.net ��������
������������� �ۿ�
���������������congenitalidiopathicnystagmus���������ط�������������������Ŵ��������������������
http://15390.dis.999120.net ��������
������ͷˮ������ˮ�� �ۿ�
������ͷˮ������ˮ��
http://15391.dis.999120.net ��������
������ �ۿ�
�����ף�������ͷ�ף�����������
http://15392.dis.999120.net ��������
ȱѪ�������� �ۿ�
ȱѪ�������䣻anteriorischemicopticneuropathy��ǰ��ȱѪ�������䣻ȱѪ��������ͷ����
http://15393.dis.999120.net ��������
������ �ۿ�
������
http://15394.dis.999120.net ��������
Leber�Ŵ��������� �ۿ�
Leber�Ŵ���������
http://15395.dis.999120.net ��������
����ή�� �ۿ�
������
http://15396.dis.999120.net ��������
���Ӳ�� �ۿ�
���Ӳ�������Ӳ��֢��������Ӳ��������
http://15397.dis.999120.net ��������
������ �ۿ�
������

http://15398.dis.999120.net ��������
������������ �ۿ�
����������������������״������������״������Boeck'ssarcoidofthelacrimalgland��Hutchinson-Boeckdiseaseofthelacrimalgland�����ٲ��������������ٲ����������������ٺ�-�����ϲ�
http://15399.dis.999120.net ��������
�����ۺ�֢ �ۿ�
�����ۺ�����autoimmuneexocrineglanddisease��exocrinopathy�������ۺ�֢�����۸���ؽ����ۺ�����������ۺ�����˹Լ�����ۺ���������������������ٲ���Gougerot-Houwesyndrome��Gougerot-Mikulicz-Sjogrensyndrome��Sjogren'sdisease��Sjogren���ۺ�������-�������ۺ�������-��
http://15400.dis.999120.net ��������
�����ܰ���Ƥ�� �ۿ�
�����ܰ���Ƥ����Mikulicz���������ܰ���Ƥ����
http://15401.dis.999120.net ��������
���ٶ��������� �ۿ�
���ٶ��������������ٻ������mixedtumorofthelacrimalgland
http://15402.dis.999120.net ��������
���ٶ������ٰ� �ۿ�
���ٶ������ٰ���alignantmixedtumor���������ٻ������multiformadenomaofthelacrimalgland
http://15403.dis.999120.net ��������
�������� �ۿ�
����������������������Բ����
http://15404.dis.999120.net ��������
������ �ۿ�
������
http://15405.dis.999120.net ��������
�������� �ۿ�
����������tumoroflacrimalcyst
http://15406.dis.999120.net ��������
���� �ۿ�
���Կ����Խ�Ĥ�ף����ۣ�����
http://15407.dis.999120.net ��������
���Կ����Խ�Ĥ�� �ۿ�
���Կ����Խ�Ĥ��
http://15408.dis.999120.net ��������
������Խ�Ĥ�� �ۿ�
������Խ�Ĥ�ף��ܲ��ۣ��ܾ��Խ�Ĥ�ף��ܾ���ŧ©��
http://15409.dis.999120.net ��������
��Ĥ��˲� �ۿ�
��Ĥ��˲�
http://15411.dis.999120.net ��������
ɳ�� �ۿ�
ɳ�ۣ�chlamyditrachomatisinfection�������Խ�Ĥ�ף�ɳ����ԭ���Ⱦ
http://15412.dis.999120.net ��������
�������Խ�Ĥ�� �ۿ�
�������Խ�Ĥ�ף��������Ĥ��
http://15413.dis.999120.net ��������
�����Գ�Ѫ�Խ�Ĥ�� �ۿ�
�����Գ�Ѫ�Խ�Ĥ�ף�acutehemorrhageconjunctivitis��acutehemorrhagicconjunctivitis��enteroviralconjunctivitis��Epidemichemorrhagicconjunct��epidemicofacutehemorrhagicconjunctivitis�������ޢ��Ĥ�ף��������Խ�Ĥ�ף����Գ�Ѫ�Խ�Ĥ�ף����������Գ�Ѫ�Խ�Ĥ�ף������Ժ�
http://15414.dis.999120.net ��������
��̬��Ӧ�Խ�Ĥ�� �ۿ�
��̬��Ӧ�Խ�Ĥ�ף���Ӧ�Խ�Ĥ��
http://15415.dis.999120.net ��������
����ͷ�Խ�Ĥ�� �ۿ�
����ͷ�Խ�Ĥ�ף�����ͷ״��Ĥ��
http://15416.dis.999120.net ��������
���������Խ�Ĥ�� �ۿ�
������Ĥ�ף�vernalkeratoconjunctivitis��������Ĥ��Ĥ�ף����������Խ�Ĥ��
http://15417.dis.999120.net ��������
����֢ �ۿ�
���ۣ����۲�������֢����Ĥ����֢
http://15418.dis.999120.net ��������
ʷ-Լ�ۺ��� �ۿ�
ʷ-Լ�ۺ�����BaaderƤ����ǻ�ף�Baader�ۺ�����pantomorphiaerythemaexudativum��Stevens-Johnson�ۺ����������������Ժ�ߣ������Դ����Ժ�ߣ��Ĥ�������ۺ������Ĥ-Ƥ��-���ۺ�����ʷ����˹-Լ��ѷ�ۺ�����ʷ-Լ�ۺ�֢��ectodermosiserosivapluriorifcialis��mucocutaneoocular
http://15419.dis.999120.net ��������
��״���� �ۿ�
��״���⣻�����ʾ�
http://15420.dis.999120.net ��������
�ۿ�����֯�� �ۿ�
�ۿ�����֯��
http://15421.dis.999120.net ��������
�ۿ�ŧ�� �ۿ�
�ۿ�ŧ�ף���ŧ��
http://15422.dis.999120.net ��������
�������� �ۿ�
��������
http://15423.dis.999120.net ��������
���Ժ����˨���Ծ����� �ۿ�
���Ժ����˨���Ծ����ף����Ժ����Ѫ˨�Ծ�����
http://15425.dis.999120.net ��������
�ۿ������ �ۿ�
�ۿ���������ۿ�ù����
http://15426.dis.999120.net ��������
�����״ϸ���� �ۿ�
�����״ϸ����������ƽϸ�����������״��Ƥϸ����
http://15427.dis.999120.net ��������
���ʰ� �ۿ�
���ʰ�
http://15428.dis.999120.net ��������
��������Ĥ���۲����� �ۿ�
��������Ĥ���۲�����
http://15429.dis.999120.net ��������
�ۿ�ת�������� �ۿ�
�ۿ�ת����������metastaticcarcinomaoffossaorbitalis
http://15430.dis.999120.net ��������
��״������۲� �ۿ�
��״������۲���endocrineinfiltrationexophthalmos��Gravesophthalmopathy��Graves�۲���malignamtexophthalmos������ͻ�ۣ�����˹�۲�����״�ٶ����۲�����״���۲���������ͻ�ۣ��ڷ����Խ�����ͻ�ۣ��ڷ������ۼ����䣻�ڷ���������ͻ��������ͻ���Լ�״����
http://15431.dis.999120.net ��������
�ۿ����Լ��� �ۿ�
�ۿ����Լ���
http://15432.dis.999120.net ��������
Τ������ѿ�� �ۿ�
Τ������ѿ�ף�Τ������ѿ�ף�Wegenergranulomatosis��Τ������ѿ�ײ���Wegener��ѿ�ף���������ѿ��
http://15433.dis.999120.net ��������
�۲�����β�ʲ� �ۿ�
�۲�����β�ʲ�
http://15434.dis.999120.net ��������
ľ�岡 �ۿ�
Kimura����angiolymphoidhyperplasiawitheosinophilia��ľ�岡����������ϸ���������ܰ���ѿ�ף�Ѫ���ܰ�����������������ϸ������
http://15435.dis.999120.net ��������
�ۿ����������� �ۿ�
�ۿ����������ԣ��ۿ����������Բ�
http://15436.dis.999120.net ��������
�ۿ�Ƥ������ �ۿ�
�ۿ�Ƥ������
http://15437.dis.999120.net ��������
�ۿ���̥�� �ۿ�
�ۿ���̥����teratomaoforbit
http://15438.dis.999120.net ��������
������С����ϲ��ۿ����� �ۿ�
������С����ϲ��ۿ����ף�microphthalmiaassociatedwithpseudogliomatosisoftheretinaandpse��С�����������Ĥ���Խ����������ۿ����Խ�������

http://15439.dis.999120.net ��������
�ۿ���Ĥ-����� �ۿ�
�ۿ���Ĥ-�����
http://15440.dis.999120.net ��������
�ۿ����������� �ۿ�
�ۿ�����������
http://15441.dis.999120.net ��������
�ۿ��Һ���� �ۿ�
�ۿ��Һ���ף��ۿ�ճҺ����
http://15442.dis.999120.net ��������
�ۿ�Ѫ�� �ۿ�
�ۿ�Ѫ��
http://15443.dis.999120.net ��������
�ۿ�Ѫ����Ƥ�� �ۿ�
�ۿ�Ѫ����Ƥ�����ۿ�Ѫ����Ƥϸ������hemangio-peritheliomaoffossaorbitalis
http://15444.dis.999120.net ��������
�۲�ëϸѪ���� �ۿ�
�۲�ëϸѪ������infantilehemangioma����ݮ�룻�۲���ɫ�룻capillaryangiomasofocularregion��capillarytumorofocularregion��infantilehemangiomaofocularregion��nevusflammeusofocularregion��Ӥ����Ѫ����
http://15445.dis.999120.net ��������
�ۿ��ں���״Ѫ���� �ۿ�
�ۿ��ں���״Ѫ����
http://15446.dis.999120.net ��������
�ۿ�������Ѫ���� �ۿ�
�ۿ�������Ѫ������venoushemangiomaoffossaorbitalis
http://15447.dis.999120.net ��������
�ۿ��ܰ��� �ۿ�
�ۿ��ܰ�����angiolymphomaoffossaorbitalis��angiomalymphaticumoffossaorbitalis���ۿ��ܰ�����
http://15448.dis.999120.net ��������
�ۿ��ھ������� �ۿ�
�ۿ��ھ�������
http://15449.dis.999120.net ��������
������������� �ۿ�
�������������
http://15450.dis.999120.net ��������
�ۿ����Ƽ����� �ۿ�
�ۿ����Ƽ�������orbitalrhabdomyosarcoma
http://15451.dis.999120.net ��������
�ۿ�ƽ������ �ۿ�
�ۿ�ƽ��������leiomyomaoffossaorbitalis
http://15452.dis.999120.net ��������
�ۿ���ά���� �ۿ�
�ۿ���ά������fibromasarcomatosumoffossaorbitalis��fibrosarcomaoffossaorbitalis
http://15453.dis.999120.net ��������
������ά��֯ϸ���� �ۿ�
������ά��֯ϸ�������ǵ�����ά��֯ϸ��������ɫ��ά������ά��ɫ����������״��ά��ɫ��
http://15454.dis.999120.net ��������
�ۿ�֬���� �ۿ�
�ۿ�֬������pimelomaliparomphalusoffossaorbitalis���ۿ�֬��
http://15455.dis.999120.net ��������
�ۿ�֬������ �ۿ�
�ۿ�֬��������adiposesarcomaoffossaorbitalis��lipoblastomaoffossaorbitalis���ۿ�֬ĸϸ�������ۿ�֬����
http://15456.dis.999120.net ��������
�ۿ����� �ۿ�
�ۿ�������osteomaorbitalis
http://15457.dis.999120.net ��������
�ۿ�����ά�쳣��ֳ֢ �ۿ�
�ۿ�����ά�쳣��ֳ֢���ۿ�����ά�Խṹ������fibrousdysplasiaoffossaorbitalis���ۿ�����ά�쳣��ֳ
http://15459.dis.999120.net ��������
�ۿ��ǻ���ά�� �ۿ�
�ۿ��ǻ���ά�����������ۿ��ǻ���ά����fossaorbitalisossifyingfibromaofbone
http://15460.dis.999120.net ��������
�������� �ۿ�
����������juvenilepilocyticastrocytoma��opticglioma����ͯ��ά����������ϸ����
http://15461.dis.999120.net ��������
��Ĥ�� �ۿ�
��Ĥ�����Լ�Ĥ����Ӳ�Լ�Ĥ������Ӳ��Ĥ����
http://15462.dis.999120.net ��������
�ۿ������� �ۿ�
�ۿ����������ۿ�ѩ��ϸ������orbitalneurinoma���ۿ�������ϸ����
http://15463.dis.999120.net ��������
�ۿ�����ά�� �ۿ�
�ۿ�����ά��
http://15464.dis.999120.net ��������
����ά���� �ۿ�
����ά������elehantiasisneuromatosa��molcuseumfibrousm��Recklinghausen����VonRecklinghausen����VonRecklinghausenDisease���������ά�����롤���ֺ�ɭ�������ֻ����������ֻ����ϲ�����ά�����ࣻ��Ƥ����������neuroinomatosis��Recklinghausen'sdisease��von
http://15465.dis.999120.net ��������
�ۿ��ǻ��������ܰ����� �ۿ�
�ۿ��ǻ��������ܰ�����
http://15466.dis.999120.net ��������
�ۿ���������ѿ�� �ۿ�
�ۿ���������ѿ�ף�fossaorbitalisLangerhanscellhistiocytosis���ۿ��ʸ�˹ϸ����֯ϸ�����ಡ���ۿ������ϸ����ѿ�ף��ۿ�����ϸ����ѿ�ף��ۿ�����ϸ������ѿ��
http://15467.dis.999120.net ��������
����-���ն�-��˼˹�ٲ� �ۿ�
����-���ն�-��˼˹�ٲ����Ǽ��Ի����Էֻ�����֯ϸ�����ಡ
http://15468.dis.999120.net ��������
������-��Τ�� �ۿ�
������-��Τ��������֬��֯ϸ�����ಡ����-�����ϲ�����-����
http://15469.dis.999120.net ��������
�����Ի�ɫ��ѿ�� �ۿ�
�����Ի�ɫ��ѿ�ף�congenitalxanthomamultiplex��nevusxanthoendothelioma�����������ѿ�ף��������ɫ��ѿ�ף������Ի�ɫ�������壻�����Ƥ��
http://15470.dis.999120.net ��������
ϸ���Խ�Ĥ�� �ۿ�
ϸ���Խ�Ĥ��
http://15471.dis.999120.net ��������
ͭ�̼ٵ������Խ�Ĥ�� �ۿ�
ͭ�̼ٵ������Խ�Ĥ�ף���ŧ�˾��Խ�Ĥ��
http://15472.dis.999120.net ��������
����Խ�Ĥ���� �ۿ�
����Խ�Ĥ����
http://15473.dis.999120.net ��������
�ǽ�˷�֦�˾��Խ�Ĥ�� �ۿ�
�ǽ�˷�֦�˾��Խ�Ĥ�ף��ǵ��ͷ�֦�˾���Ĥ�ף��ǽ���Է�֦�˾���Ĥ��
http://15474.dis.999120.net ��������
����Խ�Ĥ���� �ۿ�
����Խ�Ĥ����
http://15475.dis.999120.net ��������
÷���Խ�Ĥ���� �ۿ�
÷���Խ�Ĥ����
http://15476.dis.999120.net ��������
����Խ�Ĥ�� �ۿ�
����Խ�Ĥ��
http://15477.dis.999120.net ��������
����������Խ�Ĥ�� �ۿ�
����������Խ�Ĥ�ף����������Խ�Ĥ��
http://15478.dis.999120.net ��������
ˮ���Խ�Ĥ�� �ۿ�
ˮ���Խ�Ĥ��
http://15480.dis.999120.net ��������
�����Խ�Ĥ��Ĥ�� �ۿ�
�����Խ�Ĥ��Ĥ�ף������Խǽ�Ĥ�ף������Խ�Ĥ��Ĥ�ף������Խǽ�Ĥ��
http://15481.dis.999120.net ��������
С���Ǻ��Ს���Խ�Ĥ�� �ۿ�
С���Ǻ��Ს���Խ�Ĥ�ף������Գ�Ѫ�Խ�Ĥ��Ĥ��
http://15482.dis.999120.net ��������
�����װ��Խ�Ĥ�� �ۿ�
�����װ��Խ�Ĥ�ף������װͽ�Ĥ��


http://15483.dis.999120.net ��������
��Դ�Խ�Ĥ�� �ۿ�
��Դ�Խ�Ĥ��
http://15484.dis.999120.net ��������
Terrien��Ĥ��Ե�Ա��� �ۿ�
Terrien��Ĥ��Ե�Ա��ԣ�Terrien��Ĥ��Ե���ԣ���Ĥ�ܱ߲���״���ԣ������Խ�ĤӪ������
http://15485.dis.999120.net ��������
��״��Ĥ���� �ۿ�
��״��Ĥ���䣻�����״��Ĥ�����Ƴ����Խ�Ĥ���ƻ���״��Ĥ���䣻��Ĥ��״���ԣ���Ĥ��״����
http://15486.dis.999120.net ��������
����״��ĤӪ������ �ۿ�
����״��ĤӪ������������״��Ĥ����
http://15487.dis.999120.net ��������
Fuchs��Ĥ��ƤӪ������ �ۿ�
Fuchs��Ĥ��ƤӪ��������corneaguttata����״��Ĥ����Ĥ��״����
http://15488.dis.999120.net ��������
Reis-Bucklers��ĤӪ������ �ۿ�
Reis-Bucklers��ĤӪ������
http://15489.dis.999120.net ��������
Բ��Ĥ �ۿ�
Բ��Ĥ��Բ�ν�Ĥ
http://15490.dis.999120.net ��������
������ �ۿ�
��������Bowen����intraepithelialneoplasia��squamouscellcarcimomainsitu����ǰ�ǻ�����������ǰƤ�ף����²���Ƥ����ǰ�ڲ��䣻��Ƥ����Ƥ����ԭλ��״ϸ������Ƥ��ԭλ����intraepithelialneoplasiaofcorneaandconjunctiva���ǽ�Ĥ��Ƥ����Ƥ��
http://15491.dis.999120.net ��������
��Ĥ��״ϸ���� �ۿ�
��Ĥ��״ϸ��������Ĥ��ƽϸ��������Ĥ��״��Ƥϸ������corneal��cornealpricklecellcarcinoma��cornealsquamouscancer��cornealsquamouscarcinoma����Ĥ��ϸ��������Ĥ�۰�
http://15492.dis.999120.net ��������
���ɫ��������Ա�Ե�Խ�Ĥ�� �ۿ�
���ɫ��������Ա�Ե�Խ�Ĥ�ף���Ե�Կ����Խ�Ĥ�ף������Խ�Ĥ����
http://15493.dis.999120.net ��������
��Ĥ������ �ۿ�
��Ĥ�����ף�non-ulcerativekeratitis���������Խ�Ĥ�ף������Խ�Ĥ�ף���Ĥ�����ף�ʵ���Խ�Ĥ��
http://15494.dis.999120.net ��������
��ʳ�Խ�Ĥ���� �ۿ�
��ʳ�Խ�Ĥ����Mooren��Ĥ����
http://15495.dis.999120.net ��������
Theodore�Ϸ���ĤԵ�ǽ�Ĥ�� �ۿ�
Theodore�Ϸ���ĤԵ�ǽ�Ĥ��
http://15496.dis.999120.net ��������
Thygesondz���״��Ĥ���� �ۿ�
Thygesondz���״��Ĥ����
http://15497.dis.999120.net ��������
�������ʪ�Թؽ����Թ�Ĥ�� �ۿ�
�������ʪ�Թؽ����Թ�Ĥ��
http://15498.dis.999120.net ��������
Τ������ѿ���Թ�Ĥ�� �ۿ�
Τ������ѿ���Թ�Ĥ�ף�Wegener��ѿ���Թ�Ĥ��
http://15499.dis.999120.net ��������
�������Թ�Ĥ�� �ۿ�
�������Թ�Ĥ�ף�Behcet���Թ�Ĥ�ף������ز��Թ�Ĥ�ף��������ϲ��Թ�Ĥ��
http://15500.dis.999120.net ��������
��ϸ���������Թ�Ĥ�� �ۿ�
��ϸ���������Թ�Ĥ�ף���ϸ���Զ������Թ�Ĥ��
http://15501.dis.999120.net ��������
ϵͳ�Ժ���Ǵ��Թ�Ĥ�� �ۿ�
ϵͳ�Ժ���Ǵ��Թ�Ĥ�ף�ȫ���Ժ���Ǵ��Թ�Ĥ��
http://15502.dis.999120.net ��������
ǿֱ�Լ������Թ�Ĥ�� �ۿ�
ǿֱ�Լ������Թ�Ĥ�ף�ǿֱ�Լ����Թ�Ĥ��

http://15503.dis.999120.net ��������
�����ۺ����Թ�Ĥ�� �ۿ�
�����ۺ����Թ�Ĥ�ף�Reiter�ۺ����Թ�Ĥ��
http://15504.dis.999120.net ��������
��м���ؽ����Թ�Ĥ�� �ۿ�
��м���ؽ����Թ�Ĥ�ף�ţƤѢ�Թؽ����Թ�Ĥ��
http://15505.dis.999120.net ��������
���Գ����Թؽ�����ع�Ĥ�� �ۿ�
���Գ����Թؽ�����ع�Ĥ��
http://15506.dis.999120.net ��������
�����Զ��������Թ�Ĥ�� �ۿ�
�����Զ��������Թ�Ĥ�ף������Զ�������Թ�Ĥ��
http://15507.dis.999120.net ��������
����Զද�����Թ�Ĥ�� �ۿ�
����Զද�����Թ�Ĥ�ף�����Զ�����Ĥ���Թ�Ĥ�ף�����Զ�Զ������Թ�Ĥ��
http://15508.dis.999120.net ��������
��������ѿ����Ѫ������ع�Ĥ�� �ۿ�
��������ѿ����Ѫ������ع�Ĥ�ף�Churg-Strauss�ۺ�����ع�Ĥ�ף���Ӧ����ѿ����Ѫ������ع�Ĥ��
http://15509.dis.999120.net ��������
������������������Ը˾��Թ�Ĥ�� �ۿ�
������������������Ը˾��Թ�Ĥ�ף�����������������������Ը˾��Թ�Ĥ��
http://15510.dis.999120.net ��������
�ǵ��ͷ�֦�˾��Թ�Ĥ�� �ۿ�
�ǵ��ͷ�֦�˾��Թ�Ĥ��
http://15511.dis.999120.net ��������
����Թ�Ĥ�� �ۿ�
����Թ�Ĥ��
http://15512.dis.999120.net ��������
÷���Թ�Ĥ�� �ۿ�
÷���Թ�Ĥ��
http://15514.dis.999120.net ��������
��ķ���Թ�Ĥ�� �ۿ�
��ķ���Թ�Ĥ�ף�Lyme���Թ�Ĥ��
http://15515.dis.999120.net ��������
��״������Թ�Ĥ�� �ۿ�
��״������Թ�Ĥ�ף���״�����Թ�Ĥ��
http://15516.dis.999120.net ��������
����������Թ�Ĥ�� �ۿ�
����������Թ�Ĥ�ף����������Թ�Ĥ�ף����������Ĥ��
http://15517.dis.999120.net ��������
�����װ��Թ�Ĥ�� �ۿ�
�����װ��Թ�Ĥ��
http://15518.dis.999120.net ��������
�����岡�Թ�Ĥ�� �ۿ�
�����岡�Թ�Ĥ�ף������没�Թ�Ĥ�ף����γ没�Թ�Ĥ��
http://15519.dis.999120.net ��������
����Թ�Ĥ�� �ۿ�
����Թ�Ĥ�ף�ù���Թ�Ĥ��
http://15520.dis.999120.net ��������
������������� �ۿ�
������������ϣ����������ϣ�������صİ�����
http://15522.dis.999120.net ��������
����֢������ �ۿ�
����֢������
http://15523.dis.999120.net ��������
��л������ �ۿ�
��л�����ϣ���л������
http://15524.dis.999120.net ��������
�ж������� �ۿ�
�ж������ϣ��ж�������
http://15525.dis.999120.net ��������
���������� �ۿ�
���������ϣ�����������
http://15526.dis.999120.net ��������
���������� �ۿ�
���������ϣ����������ϣ�����������
http://15527.dis.999120.net ��������
�������� �ۿ�
�������ϣ��̷������ϣ��̷�������
http://15528.dis.999120.net ��������
��״����λ �ۿ�
��״����λ
http://15529.dis.999120.net ��������
ԭ���Լ��Աս�������� �ۿ�
ԭ���Լ��Աս�������ۣ����Գ�Ѫ�������
http://15530.dis.999120.net ��������
ԭ�������Աս�������� �ۿ�
ԭ�������Աս��������
http://15531.dis.999120.net ��������
��״������������� �ۿ�
��״������������ۣ�malignantglaucoma����������ۣ���״�������������
http://15532.dis.999120.net ��������
ԭ���Կ���������� �ۿ�
ԭ���Կ����������
http://15533.dis.999120.net ��������
������ѹ������� �ۿ�
������ѹ������ۣ�����ѹ�������
http://15534.dis.999120.net ��������
���ڹ���������� �ۿ�
���ڹ���������ۣ���������������
http://15535.dis.999120.net ��������
����ѹ֢ �ۿ�
����ѹ֢������ѹ���۸�ѹ�����ڸ�ѹ
http://15536.dis.999120.net ��������
��֢���������� �ۿ�
��֢����������
http://15537.dis.999120.net ��������
�������������� �ۿ�
��������������

http://15538.dis.999120.net ��������
ǰ����Ѫ������� �ۿ�
ǰ����Ѫ�������
http://15539.dis.999120.net ��������
ѪӰϸ��������� �ۿ�
ѪӰϸ���������
http://15540.dis.999120.net ��������
��״��������������� �ۿ�
��״��������������ۣ������������ڼ̷�������ۣ������ڰ��������������
http://15541.dis.999120.net ��������
��״����λ��������� �ۿ�
��״����λ���������
http://15542.dis.999120.net ��������
��״���ܽ�������� �ۿ�
��״���ܽ�������ۣ�lensproteinglaucoma����״�嵰���������
http://15543.dis.999120.net ��������
��״�嵰����������� �ۿ�
��״�嵰����������ۣ�glaucomainendophthalmitisphacoanaphilactica����״�����������ۣ���״������������̷��������
http://15544.dis.999120.net ��������
ҩ������������ �ۿ�
ҩ������������
http://15545.dis.999120.net ��������
��Ĥ��Ĥ��Ƥ�ۺ��� �ۿ�
��Ĥ��Ĥ��Ƥ�ۺ�������Ĥ��Ĥ��Ƥ�ۺ�֢
http://15546.dis.999120.net ��������
�����Խ�ĤӪ������ �ۿ�
�����Խ�ĤӪ��������hereditarydeepdystrophyofcornea��hereditarymesodermaldystrophy��������Ӫ����������Ĥ���������Ӫ����������Ĥ����Ŵ���Ӫ���������ڲ������Խ�Ĥ�ף��Ŵ������ĤӪ���������Ŵ�������ҶӪ������
http://15547.dis.999120.net ��������
�����ۺ��� �ۿ�
�����ۺ�����glaucomacapsulare�������ۺ�֢����Ĥ�������
http://15548.dis.999120.net ��������
ɫ��������� �ۿ�
ɫ���������
http://15549.dis.999120.net ��������
����Ѫ��������� �ۿ�
����Ѫ���������
http://15550.dis.999120.net ��������
�Ϲ�Ĥ����ѹ������������� �ۿ�
�Ϲ�Ĥ����ѹ������������ۣ��Ϲ�Ĥ����ѹ�������µ�����ۣ��Ϲ�Ĥ����ѹ��������������
http://15551.dis.999120.net ��������
�̷�����״���ۺ��˹���״���۵������ �ۿ�
�̷�����״���ۺ��˹���״���۵�����ۣ�������������̷�����ۣ��̷�����״���ۺͼپ�״����
http://15552.dis.999120.net ��������
���Խ�Ĥ��ֲ����������� �ۿ�
���Խ�Ĥ��ֲ����������ۣ����Խ�Ĥ��ֲ���̷�����ۣ��̷��ڴ��Խ�Ĥ��ֲ���������
http://15553.dis.999120.net ��������
�����弰����Ĥ����������������� �ۿ�
�����弰����Ĥ�����������������
http://15554.dis.999120.net ��������
ԭ����Ӥ��������� �ۿ�
ԭ����Ӥ��������ۣ�ԭ����Ӥ�������

http://15555.dis.999120.net ��������
ԭ����������������� �ۿ�
ԭ����������������ۣ�ԭ������������ۣ�ԭ���������������
http://15556.dis.999120.net ��������
����ɭ�Ѷ���-�����ۺ��� �ۿ�
����ɭ�Ѷ���-�����ۺ�����Axenfeld-Rieger�쳣��Axenfeld-Rieger�ۺ�����Axenfeld-Rieger�ۺ�֢������ɭ�Ѷ���-�����ۺ�֢
http://15557.dis.999120.net ��������
�˵��쳣 �ۿ�
�˵��쳣��Peter�쳣��
http://15558.dis.999120.net ��������
��������Ĥ �ۿ�
��������Ĥ�������Ժ�Ĥȱʧ
http://15559.dis.999120.net ��������
����ԭʼ����������֢ �ۿ�
����ԭʼ����������֢
http://15560.dis.999120.net ��������
����С���� �ۿ�
��������
http://15561.dis.999120.net ��������
˹����-Τ���ۺ��� �ۿ�
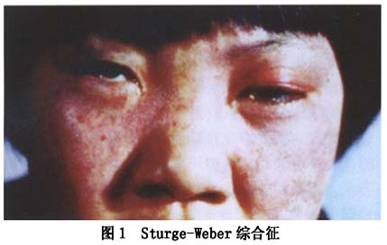
˹����-Τ���ۺ�����encephalofacialangiomatosis��Sturge-Weber�ۺ���������Ѫ����������-��Ѫ��������˹����-Τ�������ۺ�����˹����-Τ���ۺ�֢��˹-Τ�����ۺ�����encephalotrigeminalangiomatosis����������Ѫ��������ɪ־-Τ���ۺ���������Ѫ�����ۺ�������-��-Ƥ��Ѫ������
http://15562.dis.999120.net ��������
�ط���Ϣ��������ĤѪ�ܲ��� �ۿ�
�ط���Ϣ��������ĤѪ�ܲ��䣻��ɫ��Ĥ��Ѫ�ۺ���
http://15563.dis.999120.net ��������
����Ĥ��Ѫ �ۿ�
����Ĥ��Ѫ
http://15564.dis.999120.net ��������
����ĤȱѪ �ۿ�
����ĤȱѪ�����Զ�����ȱѪ������Ĥ����
http://15565.dis.999120.net ��������
��Ĥ����Ѫ�� �ۿ�
��Ĥ����Ѫ�ܣ���Ĥ����Ѫ���γɣ���Ĥ��Ѫ�ܻ�����Ĥ��Ѫ���γ�
http://15566.dis.999120.net ��������
����Ĥ����Ѫ�� �ۿ�
����Ĥ����Ѫ�ܣ�����Ĥ������Ѫ��
http://15567.dis.999120.net ��������
ǰ����Ĥ�� �ۿ�
ǰ����Ĥ�ף�ǰɫ�ز��ף�ǰ��ɫ�ز���
http://15568.dis.999120.net ��������
�۹����岡 �ۿ�
�۹����岡���۹����没���۹��γ没
http://15569.dis.999120.net ��������
�۹��׳没 �ۿ�
�۹��׳没
http://15570.dis.999120.net ��������
��β˿�没 �ۿ�
��β˿�没��blindingfilarialdisease��filarialitch��filariasisvolvulus��riverblindness����ä֢������β�߳没����ä˿�没
http://15571.dis.999120.net ��������
��������Ĥ�����ۺ��� �ۿ�
��������Ĥ�����ۺ�����Kirisawa��suveitis��������Ѫ�ܱ���������Ĥ�ף���������Ĥ�����ۺ�֢��ͩ��������Ĥ�ף��ܱ�����Ĥ��������Ѫ��ԭ�����µ�����Ĥ����
http://15572.dis.999120.net ��������
��ϸ������������Ĥ�� �ۿ�
��ϸ������������Ĥ�ף���ϸ��������ɫ�ز���
http://15573.dis.999120.net ��������
��������������Ĥ�� �ۿ�
��������������Ĥ�ף�����������ɫ�ز��ף�����������������Ĥ��
http://15574.dis.999120.net ��������
ˮ��-��״����������Ĥ�� �ۿ�
ˮ��-��״����������Ĥ�ף�ˮ����״�����������Ĥ�ף�ˮ��-��״������ɫ��Ĥ��
http://15575.dis.999120.net ��������
��������ȱ�ݲ�����������Ĥ�� �ۿ�
��������ȱ�ݲ�����������Ĥ�ף���������ȱ�ݲ�������ɫ�ز��ף�������ȱ��֢������������Ĥ��
http://15576.dis.999120.net ��������
����T�ܰ�ϸ���������µ�����Ĥ�� �ۿ�
����T�ܰ�ϸ���������µ�����Ĥ�ף�����T�ܰ�ϸ���������µ�ɫ�ز���
http://15577.dis.999120.net ��������
������Ĥ�� �ۿ�
������Ĥ�ף���ɫ�ز���
http://15578.dis.999120.net ��������
��ŧ�������� �ۿ�
��ŧ��������
http://15579.dis.999120.net ��������
�����ͯ����Ĥ�� �ۿ�
�����ͯ����Ĥ�ף������ͯɫ�ز���
http://15580.dis.999120.net ��������
���������Ĥ�� �ۿ�
���������Ĥ�ף������ɫ�ز���
http://15581.dis.999120.net ��������
÷��������Ĥ�� �ۿ�
÷��������Ĥ�ף���������������Ĥ�ף�����������ɫ��Ĥ�ף�÷����ɫ��Ĥ��
http://15582.dis.999120.net ��������
���������Ĥ�� �ۿ�
���������Ĥ�ף�Hansen��������Ĥ�ף�Hansen����ɫ��Ĥ�ף������ɫ��Ĥ��
http://15583.dis.999120.net ��������
��ķ�� �ۿ�
��ķ����Lymeborreliosis����ķ�ϲ�����ķ�������岡����ķ�������岡
http://15584.dis.999120.net ��������
�ۼ���֯���ʾ����ۺ��� �ۿ�
�ۼ���֯���ʾ����ۺ�����������֯���ʾ�����������֯���ʾ����ۺ�����������֯���ʾ����ۺ�֢������֯���ʾ����ۺ���
http://15585.dis.999120.net ��������
����ĤѪ���� �ۿ�
����ĤѪ���ף�����Ĥ������
http://15586.dis.999120.net ��������
ǿֱ�Լ�������鷢������Ĥ�� �ۿ�
ǿֱ�Լ�������鷢������Ĥ�ף�Bechterew������鷢������Ĥ�ף�Bechterew������鷢��ɫ��Ĥ�ף����ʪ�Լ�����鷢������Ĥ�ף����ʪ�Լ�����鷢��ɫ��Ĥ�ף�ǿֱ�Լ�������鷢��ɫ�ز��ף�ǿֱ�Լ�����鷢��ɫ��Ĥ��
http://15587.dis.999120.net ��������
����۽�״�����ۺ��� �ۿ�
����۽�״�����ۺ�����Posner-Schlossmansyndrome��Posner-Schlossman�ۺ�����Posner-Schlossman�ۺ�֢����-ʩ�����ۺ���������۽�״����Σ���ۺ����������-��״�����ۺ���������ۺ���
http://15588.dis.999120.net ��������
��״���������Ĥ�� �ۿ�
��״���������Ĥ�ף���״�嶾������Ĥ�ף���״�嶾��ɫ��Ĥ�ף���״�����������Ĥ�ף���״�������ɫ��Ĥ�ף���״������������ף���״�忹ԭ������Ĥ�ף���״�忹ԭ��ɫ��Ĥ�ף���״���ܽ�������Ĥ�ף���״���ܽ���ɫ��Ĥ�ף���״�������ɫ�ز��ף���״���յ�������Ĥ�ף���״��Դ������Ĥ��
http://15589.dis.999120.net ��������
���������� �ۿ�
����������
http://15590.dis.999120.net ��������
���������� �ۿ�
�������������۽�ڲ���������״����������״����
http://15591.dis.999120.net ��������
��С�ܼ�������������Ĥ���ۺ��� �ۿ�
��С�ܼ�������������Ĥ���ۺ�������С�ܼ�������������Ĥ���ۺ�֢����С�ܼ���������ɫ��Ĥ���ۺ���
http://15592.dis.999120.net ��������
���һ�����ۺ��� �ۿ�
���һ�����ۺ��������һ�����ۺ�֢���������ɢ���ۺ���
http://15593.dis.999120.net ��������
��ǹ��������Ĥ����Ĥ���� �ۿ�
��ǹ��������Ĥ����Ĥ����
http://15594.dis.999120.net ��������
���Ժ�������״ɫ����Ƥ���� �ۿ�
���Ժ�������״ɫ����Ƥ���䣻���Զ��ȱѪ������Ĥ���䣻���Ժ������״ɫ����Ƥ����
http://15595.dis.999120.net ��������
����������Ĥ�� �ۿ�
����������Ĥ�ף���ͼ״����Ĥ����Ĥ�ף�����������Ĥ����Ĥ��

http://15596.dis.999120.net ��������
��������Ĥɫ����Ƥ�� �ۿ�
��������Ĥɫ����Ƥ��
http://15597.dis.999120.net ��������
��֢�Գ�����������鷢������Ĥ�� �ۿ�
��֢�Գ�����������鷢������Ĥ�ף���֢�Գ�����������鷢��ɫ��Ĥ��
http://15598.dis.999120.net ��������
��״�ڲ�����Ĥ���� �ۿ�
��״�ڲ�����Ĥ����
http://15599.dis.999120.net ��������
����������Ĥ�װ�ȫ����Ĥ�� �ۿ�
����������Ĥ�װ�ȫ����Ĥ�ף�����������Ĥ�װ�ȫɫ�ز���
http://15600.dis.999120.net ��������
����Ĥ����ά��������Ĥ���ۺ��� �ۿ�
����Ĥ����ά��������Ĥ���ۺ���������Ĥ����ά��������Ĥ���ۺ�֢������Ĥ����ά����ɫ��Ĥ���ۺ���
http://15601.dis.999120.net ��������
˪����֦״����ĤѪ���� �ۿ�
˪����֦״����ĤѪ���ף�����������ĤѪ���ף�˪����֦״����Ĥ������Χ��
http://15602.dis.999120.net ��������
ϵͳ�Ժ���Ǵ��鷢������Ĥ�� �ۿ�
ϵͳ�Ժ���Ǵ��鷢������Ĥ�ף�ϵͳ�Ժ���Ǵ��鷢��ɫ��Ĥ��
http://15603.dis.999120.net ��������
Wegner��ѿ�װ鷢������Ĥ�� �ۿ�
Wegner��ѿ�װ鷢������Ĥ�ף�Wegner��ѿ�װ鷢��ɫ��Ĥ�ף�����������ѿ�װ鷢������Ĥ�ף�����������ѿ�װ鷢��ɫ��Ĥ��
http://15604.dis.999120.net ��������
���Ӳ���鷢������Ĥ�� �ۿ�
���Ӳ���鷢������Ĥ�ף����Ӳ���鷢��ɫ��Ĥ��
http://15605.dis.999120.net ��������
�����Զ�������װ鷢������Ĥ�� �ۿ�
�����Զ�������װ鷢������Ĥ�ף������Զ�����װ鷢������Ĥ�ף������Զ�����װ鷢��ɫ��Ĥ�ף������Զ������װ鷢������Ĥ�ף������Զ������װ鷢��ɫ��Ĥ��
http://15606.dis.999120.net ��������
����Զද���װ鷢������Ĥ�� �ۿ�
����Զද���װ鷢������Ĥ�ף�����Զද���װ鷢��ɫ��Ĥ�ף�����Զ�����װ鷢������Ĥ�ף�����Զ�����װ鷢��ɫ��Ĥ��
http://15607.dis.999120.net ��������
��м���ؽ�����鷢������Ĥ�� �ۿ�
��м���ؽ�����鷢������Ĥ�ף�ţƤѢ�Թؽ�����鷢������Ĥ�ף�ţƤѢ�Թؽ�����鷢��ɫ��Ĥ��
http://15609.dis.999120.net ��������
���������Թؽ�����鷢������Ĥ�� �ۿ�
���������Թؽ�����鷢������Ĥ�ף����������Թؽ�����鷢��ɫ��Ĥ��
http://15610.dis.999120.net ��������
�����ۺ��� �ۿ�
�����ۺ�����urethro-ocular-synovialsyndrome�����ܲ��Թؽ��ס���Ĥ�ס�����������������ض����ۺ��������ض��ۺ��������ض��ۺ������-��-��Ĥ�ۺ����������ۺ�֢��Feissiger-LeRoy-Reiter�ۺ�����infectiveuroarthitis��mucocutaneousocularsyndrome��Reiter��sDisease��Reiter
http://15611.dis.999120.net ��������
�м�����Ĥ�� �ۿ�
�м�����Ĥ�ף�parsplanitis����״��ƽ̹���ף��ܱ�����Ĥ��
http://15612.dis.999120.net ��������
������ �ۿ�
��������Behcet'sdisease��silkroutedisease���������ز��������ز����������ϲ����ڡ��ۡ���ֳ����������˿��֮·����Adamentiade��Behcet����Behcet��������Halush-Behcet�ۺ�����Touraine�ڴ����������ۺ��������������ۺ��������Բ���Ů�������Ե�Ĥ�ף�������ǰ����ŧ��

http://15613.dis.999120.net ��������
������-С��-ԭ���ۺ��� �ۿ�
������-С��-ԭ���ۺ�����Vogt-С��ԭ���ۺ�֢������Ĥ��Ĥ�����ۺ�������-ȫ����Ĥ���ۺ������ط�������Ĥ�����ף�uveoencephalitis�ۺ�����uveomeningo-encephalitis��Vogt-Koyanagi-Harada�ۺ�����Vogt-С��-ԭ���ۺ���������Ĥ�������ۺ�����ɫ��Ĥ��Ĥ���ף�С��-ԭ���ۺ�֢����-��-��-Ƥ�ۺ�
http://15614.dis.999120.net ��������
��ɫ�Ժ�Ĥ��״���� �ۿ�
��ɫ�Ժ�Ĥ��״���ף�Fuchs��Ĥ��ɫ�Ժ�Ĥ��״���ף�Fuchs����Ĥ���ۺ���
http://15615.dis.999120.net ��������
������Ĥ֢ �ۿ�
������Ĥ֢��progressivechoroidalatrophy��progressivetapetochorordalatrophy��totalchoroidalvascularatrophy��������RPE����Ĥ���ԣ�������RPEӪ�������Ա��ԣ�����������Ĥή����������̺������Ĥή����ȫ����ĤѪ��ή��
http://15616.dis.999120.net ��������
����״����Ĥ����Ĥή�� �ۿ�
����״����Ĥ����Ĥή��������Ĥ����Ĥ��״ή��
http://15617.dis.999120.net ��������
��������״����Ĥή�� �ۿ�
��������״����Ĥή����Lefletr-Wadsworth-Sidburg�ۺ������Ŵ��Իư߱���
http://15618.dis.999120.net ��������
���Խ��ӵ�����Ĥή�� �ۿ�
���Խ��ӵ�����Ĥή���������Խ��ӵ�����Ĥή��
http://15619.dis.999120.net ��������
����������Ĥή�� �ۿ�
����������Ĥή����geographichelicoidsperipapilltarychoroidopathy����ͼ״����Ĥ�ף�����������Ĥ���䣻���ܵ�ͼ״��״����Ĥ���䣻������״����Ĥ����Ĥ���б��ԣ�����������������Ĥ����Ĥ��
http://15620.dis.999120.net ��������
�۵�Ѫ�������� �ۿ�
�۵�Ѫ��������
http://15621.dis.999120.net ��������
ɫ���Ծ���������Ĥ����Ĥή�� �ۿ�
ɫ���Ծ���������Ĥ����Ĥή����������ɫ��������Ĥ����Ĥ����
http://15622.dis.999120.net ��������
��Ĥɫ���� �ۿ�
��Ĥɫ���룻��Ĥ�룻pigmentednevusofiris
http://15623.dis.999120.net ��������
��Ĥ��ɫ���� �ۿ�
��Ĥ��ɫ��������Ĥ������
http://15624.dis.999120.net ��������
��״���ɫ���� �ۿ�
��״���ɫ��������״�������
http://15625.dis.999120.net ��������
����Ĥ��ɫ���� �ۿ�
����Ĥ��ɫ����������Ĥ����
http://15626.dis.999120.net ��������
����Ĥת�ư� �ۿ�
����Ĥת�ư���metastaticcarcinomaofthechoroid������Ĥת����������Ĥת��������
http://15628.dis.999120.net ��������
����Ĥ���� �ۿ�
����Ĥ����
http://15629.dis.999120.net ��������
����Ĥ���� �ۿ�
����Ĥ����
http://15630.dis.999120.net ��������
������ ������
��������Bowen����intraepithelialneoplasia��squamouscellcarcimomainsitu����ǰ�ǻ�����������ǰƤ�ף����²���Ƥ����ǰ�ڲ��䣻��Ƥ����Ƥ����ԭλ��״ϸ������Ƥ��ԭλ����intraepithelialneoplasiaofcorneaandconjunctiva���ǽ�Ĥ��Ƥ����Ƥ��
http://15631.dis.999120.net ��������
��Ĥ��״ϸ���� ������
��Ĥ��״ϸ��������Ĥ��ƽϸ��������Ĥ��״��Ƥϸ������corneal��cornealpricklecellcarcinoma��cornealsquamouscancer��cornealsquamouscarcinoma����Ĥ��ϸ��������Ĥ�۰�
http://15632.dis.999120.net ��������
�۲�����ά���� ������
�۲�����ά������neuroinomatosisofocularregion��Recklinghausen'sdiseaseofocularregion���۲�Recklinghausen�����۲�vonRecklinghausen�����۲�vonRecklinghausen�ۺ������۲��������ά�������۲����ֻ��������۲����ֻ����ϲ�
http://15633.dis.999120.net ��������
��Ĥɫ���� ������
��Ĥɫ���룻��Ĥ�룻pigmentednevusofiris
http://15634.dis.999120.net ��������
��Ĥ��ɫ���� ������
��Ĥ��ɫ��������Ĥ������
http://15635.dis.999120.net ��������
��״���ɫ���� ������
��״���ɫ��������״�������
http://15636.dis.999120.net ��������
����Ĥ��ɫ���� ������
����Ĥ��ɫ����������Ĥ����
http://15637.dis.999120.net ��������
����Ĥת�ư� ������
����Ĥת�ư���metastaticcarcinomaofthechoroid������Ĥת����������Ĥת��������
http://15639.dis.999120.net ��������
����Ĥ���� ������
����Ĥ����
http://15640.dis.999120.net ��������
���������Ĥ������ ������
���������Ĥ������
http://15641.dis.999120.net ��������
����Ĥ������Ѫ��Ĥ ������
����Ĥ������Ѫ��Ĥ��choroidalneovascularization��CNV������Ĥ����Ѫ��Ĥ�γ�
http://15642.dis.999120.net ��������
��Ѫ������Ĥ���� ������
��Ѫ������Ĥ���䣻retinopathyduetoleucosis����ϸ����֯����֢����Ĥ���䣻Ѫ������Ĥ����
http://15643.dis.999120.net ��������
��ϸ������֢�۵� ������
��ϸ������֢�۵�
http://15644.dis.999120.net ��������
�鵰�������ϰ���ƶѪ����Ĥ���� ������
�鵰�������ϰ���ƶѪ����Ĥ���䣻���к�ƶѪ����Ĥ�������к�ƶѪ����Ĥ���䣻����ƶѪ����Ĥ���䣻�鵰�������ϰ���ƶѪ����Ĥ��
http://15645.dis.999120.net ��������
����Ĥĸϸ���� ������
����Ĥĸϸ������������Ĥϸ����������Ĥ����ϸ������retinalglioblastoma��retinalneuroblastoma��retinoma������Ĥ���Խ�����
http://15646.dis.999120.net ��������
����ĤëϸѪ��Ѫ���� ������
����ĤëϸѪ��Ѫ������vonHippel��������Ĥ��ɫ��
http://15647.dis.999120.net ��������
�ۿ���̥�� ������
�ۿ���̥����teratomaoforbit���ۿ��ڻ�̥��
http://15648.dis.999120.net ��������
�ۿ��ǻ��������ܰ��� ������
�ۿ��ǻ��������ܰ���
http://15649.dis.999120.net ��������
�����״ϸ�������۲����� ������
�����״ϸ�������۲����䣻����ƽϸ�������۲����䣻�����״��Ƥϸ�������۲�����
http://15650.dis.999120.net ��������
���ʰ����۲����� ������
���ʰ����۲�����
http://15651.dis.999120.net ��������
������Ĥ�����ۿ����� ������
������Ĥ�����ۿ����䣻�����Լ�Ĥ�����ۿ����䣻����Ӳ�Լ�Ĥ�����ۿ����䣻����Ӳ�Լ�Ĥ�������ۿ����䣻����Ӳ��Ĥ�������ۿ�����
http://15652.dis.999120.net ��������
�ۿ�ת�������� ������
�ۿ�ת����������metastaticcarcinomaoffossaorbitalis
http://15653.dis.999120.net ��������
�ۿ�Ѫ����Ƥ�� ������
�ۿ�Ѫ����Ƥ�����ۿ�Ѫ����Ƥϸ������hemangio-peritheliomaoffossaorbitalis
http://15654.dis.999120.net ��������
�۲�ëϸѪ���� ������
�۲�ëϸѪ������infantilehemangioma����ݮ�룻�۲���ɫ�룻capillaryangiomasofocularregion��capillarytumorofocularregion��infantilehemangiomaofocularregion��nevusflammeusofocularregion��Ӥ����Ѫ����
http://15655.dis.999120.net ��������
���ں���״Ѫ���� ������
���ں���״Ѫ����
http://15656.dis.999120.net ��������
�ۿ�������Ѫ���� ������
�ۿ�������Ѫ������venoushemangiomaoffossaorbitalis
http://15657.dis.999120.net ��������
�ۿ��ܰ��� ������
�ۿ��ܰ�����angiolymphomaoffossaorbitalis��angiomalymphaticumoffossaorbitalis���ۿ��ܰ�����
http://15658.dis.999120.net ��������
�ۿ����Ƽ����� ������
�ۿ����Ƽ�������orbitalrhabdomyosarcoma
http://15659.dis.999120.net ��������
�ۿ�ƽ������ ������
�ۿ�ƽ��������leiomyomaoffossaorbitalis
http://15660.dis.999120.net ��������
�ۿ���ά���� ������
�ۿ���ά������fibromasarcomatosumoffossaorbitalis��fibrosarcomaoffossaorbitalis
http://15661.dis.999120.net ��������
������ά��֯ϸ���� ������
������ά��֯ϸ�������ǵ�����ά��֯ϸ��������ɫ��ά������ά��ɫ����������״��ά��ɫ��
http://15662.dis.999120.net ��������
�ۿ�����ά�쳣��ֳ֢ ������
�ۿ�����ά�쳣��ֳ֢���ۿ�����ά�Խṹ������fibrousdysplasiaoffossaorbitalis���ۿ�����ά�쳣��ֳ
http://15663.dis.999120.net ��������
�ۿ��ǻ���ά�� ������
�ۿ��ǻ���ά�����������ۿ��ǻ���ά����fossaorbitalisossifyingfibromaofbone
http://15664.dis.999120.net ��������
�ۿ����� ������
�ۿ�������osteomaorbitalis
http://15665.dis.999120.net ��������
�������� ������
����������juvenilepilocyticastrocytoma��opticglioma����ͯ��ά����������ϸ����
http://15667.dis.999120.net ��������
��������б�����ۼ�����-������ ������
��������б�����ۼ�����-�����Σ�paraneplasticopsoclonus����������б����������
http://15668.dis.999120.net ��������
�ۿ������� ������
�ۿ����������ۿ�ѩ��ϸ������orbitalneurinoma���ۿ�������ϸ����
http://15669.dis.999120.net ��������
�ۿ�֬���� ������
�ۿ�֬������pimelomaliparomphalusoffossaorbitalis���ۿ�֬�����ۿ���֬��
http://15670.dis.999120.net ��������
�ۿ�֬������ ������
�ۿ�֬��������adiposesarcomaoffossaorbitalis��lipoblastomaoffossaorbitalis���ۿ�֬ĸϸ�������ۿ�֬����
http://15671.dis.999120.net ��������
��������ϸ���� ������
��������ϸ������Gorlin-Goltz�ۺ���
http://15672.dis.999120.net ��������
�����ٰ� ������
����Ƥ֬�ٰ��������ٰ��������ٰ���Sebaceouscarcinomaofeyelid
http://15673.dis.999120.net ��������
������������ ������
����������������������״������������״������Boeck'ssarcoidofthelacrimalgland��Hutchinson-Boeckdiseaseofthelacrimalgland�����ٲ��������������ٲ����������������ٺ�-�����ϲ�
http://15674.dis.999120.net ��������
���ٶ��������� ������
���ٶ��������������ٻ������mixedtumorofthelacrimalgland
http://15675.dis.999120.net ��������
���ٶ������ٰ� ������
���ٶ������ٰ���alignantmixedtumor���������ٻ������multiformadenomaofthelacrimalgland
http://15676.dis.999120.net ��������
�������� ������
����������������������Բ����
http://15677.dis.999120.net ��������
�������� ������
����������tumoroflacrimalcyst
http://15678.dis.999120.net ��������
����ϸ���� ������
����ϸ������astrocyticglioma��astroma����ϸ����������ϸ����
http://15679.dis.999120.net ��������
��ϵͳ�������ۺ��� ������
��ϵͳ�������ۺ�������ϵͳ�������ۺ�֢
http://15680.dis.999120.net ��������
����ĸϸ���� ������
����ĸϸ������glioblastomamultiforme��spongioblastoma���ɽ���ϸ�����������Գɽ���ϸ�����������Զ��Խ������������Խ�ĸϸ����
http://15681.dis.999120.net ��������
��֦����ϸ�����������֦����ϸ���� ������
��֦����ϸ�����������֦����ϸ��������֦�佺�����������֦�佺��������֦�������������֦����������֦����ϸ�����������֦����ϸ����
http://15682.dis.999120.net ��������
��ĸϸ���� ������
��ĸϸ����������ϸ����
http://15683.dis.999120.net ��������
�ҹ�Ĥ���� ������
�ҹ�Ĥ������ependymocytoma���ҹ�Ĥ�����ҹ�Ĥϸ����
http://15684.dis.999120.net ��������
�������ͷ״�� ������
�������ͷ״����choroidplexuspapilloma
http://15685.dis.999120.net ��������
�ɹ����� ������
�ɹ�������pinealtumor��pineocytoma
http://15686.dis.999120.net ��������
�ɹ���ϸ���� ������
�ɹ���ϸ����
http://15687.dis.999120.net ��������
������ϸ���� ������
������ϸ����
http://15688.dis.999120.net ��������
��Ԫ��������Ԫ�����ʻ�������� ������
��Ԫ��������Ԫ�����ʻ��������
http://15689.dis.999120.net ��������
�ǹ��� ������
�ǹ���
http://15690.dis.999120.net ��������
�Ǿ�ϸ���� ������
�Ǿ�ϸ���������ƹ�ϸ����
http://15691.dis.999120.net ��������
����ά�쳣����֢ ������
����ά�쳣����֢���ǹ���ά�ṹ����������ά�쳣�������Ǹ�������ά���������Ը�������ά��
http://15692.dis.999120.net ��������
��������Ĥ�� ������
��������Ĥ����cerebralconvexitymeningima
http://15693.dis.999120.net ��������
ʸ״�����Ĥ�� ������
ʸ״�����Ĥ������ʸ״����Ĥ��
http://15694.dis.999120.net ��������
����������Ĥ�� ������
����������Ĥ����������Ĥ�������������Լ�Ĥ������������Ӳ�Լ�Ĥ��������������Ӳ��Ĥ����
http://15695.dis.999120.net ��������
�����Ĥ�� ������
�����Ĥ��������Լ�Ĥ�����Լ�Ĥ�������Լ�Ĥ��ά��������Ĥ��������Ĥ��ά�����������Ĥ����
http://15697.dis.999120.net ��������
��������Ĥ�� ������
��������Ĥ��
http://15698.dis.999120.net ��������
�������Ĥ�� ������
�������Ĥ����������Ĥ��

http://15699.dis.999120.net ��������
�ṵ��Ĥ�� ������
�ṵ��Ĥ��
http://15700.dis.999120.net ��������
������Ĥ�� ������
������Ĥ����meningiomaofmiddlefossa�������Լ�Ĥ����������Ĥ��������Ӳ�Լ�)Ĥ��������Ӳ��Ĥ����
http://15701.dis.999120.net ��������
С�����Ž���Ĥ�� ������
С�����Ž���Ĥ����meningiomaofcerebellopontineangle����С�Խ���Ĥ��
http://15702.dis.999120.net ��������
�ҹ�б����Ĥ�� ������
�ҹ�б����Ĥ����slopedmeningiomaofpetrousbone
http://15704.dis.999120.net ��������
���Ǵ����Ĥ�� ������
���Ǵ����Ĥ����meningiomaoftheforamenoccipitablemagnum���������Ĥ�������Ǵ������Ĥ��
http://15705.dis.999120.net ��������
��Ĥ���� ������
��Ĥ������meningosarcoma
http://15706.dis.999120.net ��������
������Ĥ�� ������
������Ĥ������Ĥ��������
http://15707.dis.999120.net ��������
��Ĥ���� ������
��Ĥ�������Լ�Ĥ����
http://15708.dis.999120.net ��������
�������� ������
����������hypophysealadenoma�����´�������
http://15709.dis.999120.net ��������
�������� ������
����������prolactin-producingtumor�������ط���������������
http://15710.dis.999120.net ��������
���ܴ������� ������
���ܴ���������clinicallyinactivepituitaryadenoma��endocrineinactiveadenoma��functionlesspituitaryadenoma��nonsecretorypituitaryadenoma���Ƿ����Դ����������ٴ����ܴ����������ٴ����Դ������������ܴ������������ڷ��ڻ���������nonfunctioninghypophysealadenom
http://15711.dis.999120.net ��������
�ʹ��� ������
�ʹ������������ף�����������������������������������ʹ�����������Ƥ���ף�������������ϸ������craniopharyngealducttumor��hypophyseal-ducttumor��Rathke'stumor��suprasellarcyst�������º�ķ������Ƥ��������������Լ���������������������������ף����ؿ�������
http://15712.dis.999120.net ��������
��Ƥ���� ������
��Ƥ���ף�epidermoidcyst����Ƥ�����ף�intracranialepidermalcyst�����ڱ�Ƥ�����ף�������Ƥ�������������������������Ե�֬��
http://15713.dis.999120.net ��������
Ƥ������ ������
Ƥ�����ף�dermoidtumor��Ƥ������Ƥ��������intracranialdermoidcyst��intracranialdermoidtumor��zoomylus������Ƥ����������Ƥ�����ף�����Ƥ������
http://15714.dis.999120.net ��������
�ڼ����� ������
�ڼ����������ڼ�����
http://15715.dis.999120.net ��������
�������� ������
����������acousticnerveneurilemmoma��acousticneurinoma
http://15716.dis.999120.net ��������
����ֳϸ���� ������
����ֳϸ�������ǵ��ͻ�̥����������ֳϸ����������ֳϸ����������֯����intracranialgermcelltumor��intracranialgermcelltumour��intracranialgerminaltumor��intracranialgonioma��������֯�������ڷǵ��ͻ�̥������������ֳϸ��������������֯���������ɹ�����
http://15717.dis.999120.net ��������
�ڻ�̥�� ������
�ڻ�̥�������ڻ�̥��
http://15718.dis.999120.net ��������
�Ը����� ������
�Ը�������brainstemtumor
http://15719.dis.999120.net ��������
�ڶ����� ������
�ڶ�������congenitalcerebralaneurysm�����ڶ��������������Զ�����
http://15720.dis.999120.net ��������
�ں���״Ѫ���� ������
�ں���״Ѫ����������״Ѫ�ܻ��Σ����ں���״Ѫ����
http://15721.dis.999120.net ��������
���Դ����� ������
���Դ����������Դ�����������Ѫ�ܻ��Σ����Դ������Σ����Դ������ţ����Դ�����
http://15722.dis.999120.net ��������
�������Գ�Ѫ ������
�������Գ�Ѫ����������Ѫ
http://15723.dis.999120.net ��������
Ѫ����֯ϸ���� ������
Ѫ����֯ϸ������ëϸѪ��Ѫ������Ѫ��ĸϸ������Ѫ����Ƥ����Ѫ����Ƥ����Ѫ����״��Ƥ����Lindau��������ëϸѪ��Ѫ����������Ѫ��ĸϸ����������Ѫ����Ƥ��������Ѫ����Ƥ��������Ѫ����֯ϸ����������Ѫ����״��Ƥ��������Ѫ����״ϸ����
http://15724.dis.999120.net ��������
���������� ������
������������glomusjugularetumor��tumoroftheglomusjugularis�����ȸ��Ը�������������ѧ�����������ྱ��������
http://15725.dis.999120.net ��������
������ϵͳ�ܰ��� ������
������ϵͳ�ܰ�����������ϵͳ�����ܰ���
http://15726.dis.999120.net ��������
��ת���� ������
��ת��������ת��������������ת����������ת��������
http://15727.dis.999120.net ��������
�ں�ɫ���� ������
�ں�ɫ�������ں����������ں�ɫ���������ں�����
http://15728.dis.999120.net ��������
��֬���� ������
��֬������intracranialliparomphalus��intracranialpimeloma������֬����
http://15729.dis.999120.net ��������
�������Ա�Եϵͳ���� ������
�������Ա�Եϵͳ���ף��������Ա�Եϵ���ף��������Ա�Եϵ���ף��������Ա�Եϵͳ����
http://15730.dis.999120.net ��������
��������С�Ա��� ������
��������С�Ա���
http://15731.dis.999120.net ��������
��ɫ���� ������
��ɫ��������ɫ��ϸ������������
http://15732.dis.999120.net ��������
ͷ����״ϸ���� ������
ͷ����״ϸ������cephalicepidermoidcarcinoma��cephalicpricklecellcarcinoma��cephalicsquamouscancer��cephalicsquamouscellcarcinoma��ͷ����ƽϸ������ͷ����Ƥ������ͷ����ϸ������ͷ���۰���ͷ����״��Ƥϸ����
http://15733.dis.999120.net ��������
�������鷢�ľ����ϰ� ������
�������鷢�ľ����ϰ����������鷢�ľ����������鷢�ľ�����ң��������鷢�ľ�������
http://15734.dis.999120.net ��������
��֢�鷢�ľ����ϰ� ������
��֢�鷢�ľ����ϰ�����֢�鷢�ľ�����֢�鷢�ľ�����ң���֢�鷢�ľ�������
http://15735.dis.999120.net ��������
��ǻ�˻�״��ͷ���� ������
��ǻ�˻�״��ͷ��������ǻ�˻�����ͷ����
http://15736.dis.999120.net ��������
�����˷ΰ� ������
�����˷ΰ���senilelungcarcinoma��senilepulmonarycarcinoma��senilepulmonarycarcinosis������ΰ�
http://15737.dis.999120.net ��������
�����˸��������� ������
�����˸�����������senileabdominalaneurysm�����긹��������
http://15738.dis.999120.net ��������
�����˼�״�ٰ� ������
�����˼�״�ٰ���senilestrumamaligna��senilethyroidcancer��senilethyroidcarcinoma�������״�ٰ��������˶��Լ�״���ף�������Լ�״����
http://15739.dis.999120.net ��������
���������� ������
������������senilecarcinomaofkidney��������������������ϸ���������������ٰ�����������ϸ������������ϸ�������������ٰ���������ϸ����
http://15740.dis.999120.net ��������
�����˰��װ� ������
�����˰��װ���senilecarcinomaofurinarybladder��������װ�
http://15741.dis.999120.net ��������
�����˵��Ұ� ������
�����˵��Ұ������굨�Ұ�
http://15742.dis.999120.net ��������
�����˴����� ������
�����˴�������senilehypophysoma��senilepituitarytumor��seniletumorofhypophysis�����괹�����������˴�������
http://15743.dis.999120.net ��������
���갩֢���˵ļ��Ը�Ⱦ ������
���갩֢���˵ļ��Ը�Ⱦ�����갩֢���˼��Ը�Ⱦ
http://15745.dis.999120.net ��������
�����˼����ܰ�ϸ����Ѫ�� ������
�����˼����ܰ�ϸ����Ѫ����senileacutelympoidleukemia�����꼱���ܰ�ϸ����Ѫ��
http://15746.dis.999120.net ��������
�����˼��Է��ܰ�ϸ����Ѫ�� ������
�����˼��Է��ܰ�ϸ����Ѫ����senileacutenon-lympoidleukemia�����꼱�Է��ܰ�ϸ����Ѫ��
http://15747.dis.999120.net ��������
���꼱��Ѫ�� ������
���꼱��Ѫ����senileacuteleucemia
http://15748.dis.999120.net ��������
������������ϸ����Ѫ�� ������
������������ϸ����Ѫ����senilechronicgranulocyticleukemia������������ϸ����Ѫ����������������ϸ����Ѫ����������������ϸ����Ѫ��
http://15749.dis.999120.net ��������
�����������ܰ�ϸ����Ѫ�� ������
�����������ܰ�ϸ����Ѫ����senilechroniclymphocyticleukemia�����������ܰ�ϸ����Ѫ���������������ܰ�ϸ����Ѫ��
http://15750.dis.999120.net ��������
������� ������
������θ����senilecarcinomaofstomach��senilegastriccarcinoma��������θ�ٰ�������θ��
http://15751.dis.999120.net ��������
�����˻�����ܰ��� ������
�����˻�����ܰ�����senileHodgkin��slymphoma�����������ܰ���
http://15752.dis.999120.net ��������
�����˷ǻ��������ܰ����� ������
�����˷ǻ��������ܰ�����������ǻ�����ܰ���
http://15753.dis.999120.net ��������
�����˶�Թ����� ������
�����˶�Թ������������Թ������������˺����ز��������˺������ϲ���senileHuppert'sdisease����������ز�������������ϲ�
http://15754.dis.999120.net ��������
������ǰ���ٰ� ������
������ǰ���ٰ���senileprostaticcarcinoma������ǰ���ٰ�
http://15755.dis.999120.net ��������
������ԭ���Ըΰ� ������
������ԭ���Ըΰ���senileprimaryhepaticcarcinoma������ԭ���Ըΰ�
http://15756.dis.999120.net ��������
�����˴����� ������
�����˴�������senilecarcinomaoflargeintestine�����������
http://15757.dis.999120.net ��������
������ʳ�ܰ� ������
������ʳ�ܰ���senileesophagealcarcinoma������ʳ�ܰ�
http://15758.dis.999120.net ��������
���������ٰ� ������
���������ٰ���senilecancerofpancreas��senilepancreaticcancer���������ٰ�
http://15759.dis.999120.net ��������
���������ٰ� ������
���������ٰ���senilebreastcancer��senilemammaryadenocarcinoma��senilemammarycancer���������鰩�����������ڣ��������ٰ�
http://15760.dis.999120.net ��������
�������������� ������
����������������������������
http://15761.dis.999120.net ��������
���ٻ���� ������
���ٻ������Pleomorphicadenoma�����ٶ���������
http://15762.dis.999120.net ��������
����Ѫ���� ������
����Ѫ������cervicalangioma
http://15763.dis.999120.net ��������
���ٶ������� ������
���ٶ������������ٶ����������ٶ�����
http://15764.dis.999120.net ��������
�����ܰͽ�ת�ư� ������
�����ܰͽ�ת�ư�
http://15765.dis.999120.net ��������
��״���� ������
��״����������״����
http://15766.dis.999120.net ��������
�����Ը߹����Լ�״������ ������
�����Ը߹����Լ�״��������toxicadenoma�����������������Թ��ܿ����Լ�״������
http://15767.dis.999120.net ��������
��״�ٰ� ������
��״�ٰ���strumamaligna��thyroidcancer�����Լ�״����
http://15768.dis.999120.net ��������
��״��С�� ������
��״��С������״����������thyroidminimumcancer
http://15769.dis.999120.net ��������
�����Լ�״�ٷ������� ������
�����Լ�״�ٷ�������
http://15770.dis.999120.net ��������
ԭ���Լ�״�ٶ����ܰ��� ������
ԭ���Լ�״�ٶ����ܰ�����ԭ���Լ�״�ٺνܽ�ԭ���Լ�״�ٺνܽ��ϲ�
http://15771.dis.999120.net ��������
��ת�������� ������
��ת�����������̷��Է�����
http://15773.dis.999120.net ��������
֧������������ѿ�ײ� ������
֧������������ѿ�ײ�
http://15774.dis.999120.net ��������
��������� ������
�����������pulmonarybenigntumors������������
http://15775.dis.999120.net ��������
������Ѫ������ѿ�ײ� ������
������Ѫ������ѿ�ײ���allergicgranulomatosis��Churg-Strauss�ۺ�������̬��Ӧ����ѿ�ײ�����Ӧ����ѿ�ף���Ӧ����ѿ�ײ�����������ѿ�ף���������ѿ����Ѫ���ף���������ѿ��֢��Churg-Strausssvndrome��allergicangiitisandgranulomatosis��Churg-Strausesyndrome��allergicgranulomatou
http://15776.dis.999120.net ��������
�������ܰ�ϸ��Ѫ������ѿ�ײ� ������
�������ܰ�ϸ��Ѫ������ѿ�ײ�
http://15777.dis.999120.net ��������
�μ����ܰ��� ������
�μ����ܰ�����nodularlymphoidhyperplasia�����ܰ�����������ܰ���֯���������μ��ܰ�����������ܰ���֯����
http://15778.dis.999120.net ��������
�ΰ� ������
������������
http://15779.dis.999120.net ��������
�β��ټ��Ķ������� ������
�β��ټ��Ķ����������β��ټ��Ķ���������β��ټ��Ķ�������
http://15780.dis.999120.net ��������
������ ������
������
http://15781.dis.999120.net ��������
���ٰ� ������
���ٰ�
http://15782.dis.999120.net ��������
����� ������
�����
http://15783.dis.999120.net ��������
����Сϸ���� ������
����Сϸ��������������ϸ������thymicoatcellcarcinoma��thymicsmallcellcancer��thymicsmall-cellcarcinoma������������ϸ����
http://15784.dis.999120.net ��������
�ݸ�������ܰ��� ������
�ݸ�������ܰ������ݸ��νܽ��ݸ��ܰ���ѿ�ף��ݸ���-˹����hodgkinlymphomaofmediastinum
http://15785.dis.999120.net ��������
�ݸ��ǻ�����ܰ��� ������
�ݸ��ǻ�����ܰ������ݸ��Ǻνܽ��ܰ���
http://15786.dis.999120.net ��������
�ݸ�����Դ������ ������
�ݸ�����Դ������
http://15787.dis.999120.net ��������
ֲ����ϵͳ���� ������
ֲ����ϵͳ������������ϵ�������ݸ�������ϵ����
http://15788.dis.999120.net ��������
�ݸ�������Դ������ ������
�ݸ�������Դ������
http://15789.dis.999120.net ��������
�ݸ���ҶԴ���������������� ������
�ݸ���ҶԴ����������������
http://15790.dis.999120.net ��������
ʳ������ ������
ʳ������
http://15791.dis.999120.net ��������
ʳ��ƽ������ ������
ʳ��ƽ������
http://15792.dis.999120.net ��������
ʳ����ͷ״�� ������
ʳ����ͷ״��
http://15793.dis.999120.net ��������
ʳ��Ѫ���� ������
ʳ��Ѫ������solitaryphlebectasis���Ĥ��������ʳ��Ѫ������ʳ�ܹ����Ծ������ţ�esophagushemangioma
http://15794.dis.999120.net ��������
ʳ���� ������
ʳ�ܰ�����֢��ʳ������ҭ����carcinomaofesophagus
http://15795.dis.999120.net ��������
���� ������
������������
http://15796.dis.999120.net ��������
ԭ����ʳ�ܶ��Ժ�ɫ���� ������
ԭ����ʳ�ܶ��Ժ�ɫ������ԭ����ʳ�ܶ��Ժ�����
http://15798.dis.999120.net ��������
ʳ��� ������
ʳ�����ʳ�����
http://15799.dis.999120.net ��������
ԭ����ʳ�ܶ����ܰ��� ������
ԭ����ʳ�ܶ����ܰ���
http://15800.dis.999120.net ��������
�����Һ�� ������
�����Һ��������ճҺ��

http://15801.dis.999120.net ��������
������Ƽ��� ������
������Ƽ�����cardiacrhabdomyoma��hamartmaofheart��histiocyticcardiomyopathy��Purkinjeϸ�������������������֯ϸ�����ļ���
http://15802.dis.999120.net ��������
�Ҽ��Ĥ���� ������
�Ҽ��Ĥ����
http://15803.dis.999120.net ��������
����֬���� ������
����֬������liparomphalusofheart��pimelomaofheart������֬��
http://15804.dis.999120.net ��������
������ͷ��������ά�� ������
������ͷ��������ά����GiantLamblia��sexcrescenceofheart������Ĥ���������Ĥ��ͷ״�������൯����ά������������������ࣻ������ͷ����������ͷ״������ά����������ͷ״��ά��
http://15805.dis.999120.net ��������
���ķ�����ǻ����ƽ������ ������
���ķ�����ǻ����ƽ������
http://15806.dis.999120.net ��������
�����Һ�� ������
�����Һ��������ճҺ��
http://15807.dis.999120.net ��������
�����Ҷ���ʩ���� ������
�����Ҷ���ʩ������malignantperipheralgliomaofleftventricle�������Ҷ���Schwann���������Ҷ�����Ĥ���������Ҷ����������������Ҷ���������������������ά��������������Դ������
http://15808.dis.999120.net ��������
ԭ����������Ƽ����� ������
ԭ����������Ƽ�������primaryrhabdosarcomaofheart
http://15809.dis.999120.net ��������
����ת���� ������
����ת����������ת��������
http://15810.dis.999120.net ��������
����ת���Զ��Ժ�ɫ���� ������
����ת���Զ��Ժ�ɫ����
http://15811.dis.999120.net ��������
�İ��ڶ��Ի�̥�� ������
�İ��ڶ��Ի�̥���������İ���̥��
http://15812.dis.999120.net ��������
�İ���Ƥ�� ������
�İ���Ƥ����celiotheliomaofpericardium��celotheliomaofpericardium��mesohylomaofpericardium
http://15813.dis.999120.net ��������
�������� ������
��������
http://15814.dis.999120.net ��������
�İ�ת���� ������
�İ�ת�������İ�ת��������
http://15815.dis.999120.net ��������
��������Ĥ��Ƥ�� ������
��������Ĥ��Ƥ���������ͼ�Ƥ�������Լ�Ƥ������Ĥ��������ά����localizedpleuralendotheliomas�������Լ�Ƥ��
http://15816.dis.999120.net ��������
���Լ�Ƥ�� ������
��������Ĥ��Ƥ�������Լ�Ƥ����diffusepleuralendotheliomas
http://15817.dis.999120.net ��������
�ر�����֯���� ������
�ر�����֯����

http://15820.dis.999120.net ��������
��ͷ����ͷ״�� ������
��ͷ����ͷ״����papillaryepitheliomaofpapilla����ͷ����ë״������ͷ����ͷ������ͷ����ͷ״��Ƥ��
http://15822.dis.999120.net ��������
�鷿��������ͷ״�� ������
�鷿��������ͷ״�����鷿����ͷ״�����鷿������ͷ״��������
http://15823.dis.999120.net ��������
���������� ������
������ά������������������ά���������Һ��ά��������������
http://15824.dis.999120.net ��������
���ٴ����� ������
���ٴ�������������������hamartomaoflactealgland
http://15825.dis.999120.net ��������
�鷿����ά�� ������
�鷿����ά�����鷿��ά������fibroneuromaofbreast��neuroinomaofbreast
http://15826.dis.999120.net ��������
���ٰ� ������
���ٰ���breastcancer��mammaryadenocarcinoma
http://15827.dis.999120.net ��������
�������ٰ� ������
�������ٰ���earlybreastcancer��earlymammaryadenocarcinoma
http://15828.dis.999120.net ��������
����ʪ������ ������
����ʪ������������Paget����������Ƥ����ʪ�Paget'sdiseaseofbreast
http://15829.dis.999120.net ��������
�������ٰ� ������
�������ٰ������������ף����������ٰ����������ٰ���������������������Ƥ�ܰͰ������������ٰ�
http://15830.dis.999120.net ��������
�鷿��ʪ������ ������
�鷿��ʪ��������extramammaryPaget'sdisease���鷿��Paget�����������弪�ز����������弪���ϲ�
http://15833.dis.999120.net ��������
�������ٰ� ������
�������ٰ���malebreastcarcinoma�������鰩
http://15834.dis.999120.net ��������
����Ҷ״������ ������
����Ҷ״������������Ҷ״��������������Ҷ״����
http://15835.dis.999120.net ��������
����֬������ ������
����֬������������֬������adiposesarcomaofbreast��lipoblastomaofbreast���鷿��֬ϸ�������鷿֬ĸϸ����
http://15838.dis.999120.net ��������
�����¶���-Ҹ������ ������
�����¶���-Ҹ������
http://15840.dis.999120.net ��������
���������� ������
������������thoracicaorticaneurysms
http://15842.dis.999120.net ��������
�ظ������������� ������
�ظ���������������thoracoabdomnalaorticaneurysms���ظ���������
http://15843.dis.999120.net ��������
�������� ������
��������
http://15844.dis.999120.net ��������
���������� ������
������������intraspinalneurinoma������������ϸ����������������������ά������������ϸ����
http://15845.dis.999120.net ��������
���ڼ�Ĥ�� ������
���ڼ�Ĥ��
http://15846.dis.999120.net ��������
����ת�������� ������
����ת������������ת������������ת����
http://15847.dis.999120.net ��������
�������Լ��財 ������
�������Լ��財���������Լ��財
http://15848.dis.999120.net ��������
θƽ������ ������
θƽ������
http://15849.dis.999120.net ��������
����� ������
�������fibroneuromaofstomach��neuroinomaofstomach
http://15850.dis.999120.net ��������
θ֬���� ������
θ֬������pimelomaofstomach
http://15851.dis.999120.net ��������
θ��̥�� ������
θ��̥����dermoidtumorofstomach��dysembryomaofstomach��teratoidtumorofstomach��teratomadermoidtumorofstomach
http://15852.dis.999120.net ��������
��θ�ܰ��� ������
��θ�ܰ���
http://15853.dis.999120.net ��������
� ������
���carcinomaofstomach��stomachcancer�����������
http://15854.dis.999120.net ��������
θ�����ܰ��� ������
θ�����ܰ�����gastricmalignantlymphoma��θ�νܽ�θ�νܽ��ϲ���θ�ܰ�����ԭ����θ�ܰ���
http://15855.dis.999120.net ��������
θƽ�������� ������
θƽ����������gastricleiomyosarcoma
http://15856.dis.999120.net ��������
�� ������
����carcinoidtumorofstomach
http://15857.dis.999120.net ��������
����� ������
θ��������Zollinger-Ellisonsyndrome����θҺ���������ٷǦ�ϸ��������Դ������ԭ����θ��������֢��-���ۺ�������-�������ۺ�����gastrinadenoma��Zollinger-Ellison�ۺ���
http://15858.dis.999120.net ��������
��θƽ�������� ������
��θƽ��������
http://15859.dis.999120.net ��������
�����ȸ�ϸ���� ������
�����ȸ�ϸ����
http://15860.dis.999120.net ��������
����ƽ������ ������
����ƽ������������ƽ������������
http://15861.dis.999120.net ��������
��������ϸ���� ������
��������ϸ����
http://15862.dis.999120.net ��������
�����ٰ� ������
�����ٰ���mucinouscarcinomaofbladder�����������������Һ�ٰ�������ӡ��ϸ����
http://15863.dis.999120.net ��������
������״ϸ���� ������
������״ϸ������epidermoidcarcinomaofbladder�����ױ�Ƥ����������ϸ���������ǻ����������۰�
http://15864.dis.999120.net ��������
����ƽ�������� ������
����ƽ��������������ƽ������������
http://15865.dis.999120.net ��������
�������� ������
��������
http://15866.dis.999120.net ��������
ǰ���ٰ� ������
ǰ���ٰ���prostaticcarcinoma��tumorofprostate
http://15867.dis.999120.net ��������
������ ������
����������������ֳ������
http://15869.dis.999120.net ��������
غ������ ������
غ��������orcele��testiculoma��غ��ͻ����������������ϸ������������
http://15870.dis.999120.net ��������
������������ ������
��������������malignancyofspermaticcord��malignanttumorofspermaticcord��malignanttumourofspermaticcord���������������������������
http://15871.dis.999120.net ��������
���Ҷ������� ������
���Ҷ���������malignancyofseminalvesicle��malignanttumorofseminalvesicle��malignanttumourofseminalvesicle�����Ҷ����������Ҷ�����������Ҷ�������
http://15872.dis.999120.net ��������
���Ұ� ������
���Ұ���squamouscellcarcinomaofscrotum��������״ϸ������scroticarcinoma�����ұ�ƽϸ������������״��Ƥϸ����
http://15873.dis.999120.net ��������
�������� ������
����������eczematoidcancerofthescrotum��Paget��sdiseaseofthescrotum������Paget��������ʪ������
http://15874.dis.999120.net ��������
������ͷ״�� ������
������ͷ״����vulvarpapillaryepithelioma��������ë״����������ͷ����������ͷ״��Ƥ��
http://15875.dis.999120.net ��������
���������� ������
������������Schwannomaofvulva������ʩ��ϸ����������������ϸ������myoschwannomaofvulva��neurilemmomaofvulva��neurilemomaofvulva��peripheralgliomaofvulva��schwannogliomaofvulva��sheathtumorofvulva����������������
http://15876.dis.999120.net ��������
���������� ������
������������������������fibroneuromaofvulva��neuroinomaofvulva
http://15877.dis.999120.net ��������
���������� ������
������������������������������������
http://15878.dis.999120.net ��������
���������� ������
������������������״���������������ܺ�����������������������������ֳ��������
http://15879.dis.999120.net ��������
������������� ������
�������������������Bowen�����������������ɫ����Bowen����������Bowen�����������������������ֳ��������Bowen����������ֳ��������Bow-en��
http://15880.dis.999120.net ��������
������Ƥ������ ������
������Ƥ������

http://15881.dis.999120.net ��������
���������� ������
��������ά��������Ƥ�������������ࣻ����������ά����������ά��Ƥ������Ϣ�⣻������ά��Ƥ��Ϣ�⣻fibromamolleofvulva
http://15882.dis.999120.net ��������
����ɫ���� ������
����ɫ���룻pigmentedmoleofvulva��pigmentednevusofvulva
http://15883.dis.999120.net ��������
�������� ������
������ά����vulvardesmoma��vulvarfibromadurum��vulvarhardfibroma������Ӳ��ά����vulvarfibroidtumor��vulvarinoma
http://15884.dis.999120.net ��������
����֬���� ������
����֬������vulvarliparomphalus��vulvarpimeloma������֬��
http://15885.dis.999120.net ��������
����Ѫ���� ������
����Ѫ������vulvarangeioma��vulvarangioma��vulvarangioneoplasm��vulvarhemangioma��vulvarhemartoma��vulvarvasculartumor
http://15886.dis.999120.net ��������
�����ܰ��� ������
�����ܰ�����vulvarangiolymphoma��vulvarangiomalymphaticum�������ܰ�����
http://15887.dis.999120.net ��������
����ƽ������ ������
����ƽ��������vulvarliomyoma��vulvarmyomalevicellulare
http://15888.dis.999120.net ��������
���������Լ�ĸϸ���� ������
���������Լ�ĸϸ��������������ϸ���ɼ�ϸ������������ϸ���Գɼ�ϸ�������������Գɼ�ϸ�������������Լ�ĸϸ����
http://15889.dis.999120.net ��������
�������ڽ������۰� ������
�������ڽ������۰���superficialinvasivecarcinomaofvulvar������dz���������۰�������������״��Ƥ����
http://15890.dis.999120.net ��������
�����������۰� ������
�����������۰���������״��Ƥ����
http://15891.dis.999120.net ��������
������״�� ������
������״����verrucacarcinomaofvulva
http://15892.dis.999120.net ��������
��������ϸ���� ������
��������ϸ������basalcellepitheliomaofvulva��basalomaofvulva��basilomaofvulva��carcinomabasocellulareofvulva
http://15893.dis.999120.net ��������
�����弪�ز� ������
�����弪�ز���Ů���弪�ز�������Paget��������ʪ��������Paget'sdiseaseofvulva���ɽܲ��������弪���ϲ�
http://15894.dis.999120.net ��������
ǰͥ���ٰ� ������
ǰͥ���ٰ����Ͷ������ٰ����Ͷ����ٰ���Bartholinglandcarcinoma
http://15895.dis.999120.net ��������
������ٰ� ������
������ٰ���carcinomaoffemaleprostate��carcinomaofglandulaeparaurethrales
http://15896.dis.999120.net ��������
���������ٰ� ������
���������ٰ���sweetglandcarcinomaofvulva
http://15897.dis.999120.net ��������
�������۰� ������
�������۰��������۰�
http://15898.dis.999120.net ��������
����ƽ�������� ������
����ƽ����������liomyosarcomaofvulva
http://15899.dis.999120.net ��������
�������������� ������
������������������������ʩ������������������������������ά����
http://15900.dis.999120.net ��������
�������������� ������
��������������������Kaposi��������������ط��Գ�Ѫ�����������������Գ����Գ�Ѫ����������������������������������������������������������������Ƥ����Գ�Ѫ������
http://15901.dis.999120.net ��������
��������״����֯���� ������
��������״����֯������������״����֯��������������֯����״����
http://15902.dis.999120.net ��������
������Ĥ���� ������
������Ĥ������malignantsynoviomaofvulva��synoviosarcomaofvulva���������Ի�Ĥ��
http://15903.dis.999120.net ��������
������������ ������
������������������������������������������
http://15904.dis.999120.net ��������
����Ѫ����Ƥϸ���� ������
����Ѫ����Ƥϸ������haemangiopericytomaofvulva��peritheliomaofvulva��������Ƥ��
http://15905.dis.999120.net ��������
����ת���� ������
����ת����������ת�ư�
http://15906.dis.999120.net ��������
������ɫ���� ������
������ɫ����������������
http://15907.dis.999120.net ��������
�����ѻ����� ������
�����ѻ�������endodermalsinustumorofvulva���������߲�����������������
http://15908.dis.999120.net ��������
����÷�˶�ϸ���� ������
����÷�˶�ϸ����������Merkelϸ����������Ƥ��APUD��������Ƥ��С�������������ڷ��ڰ�������ԭ����Сϸ������������״��
http://15909.dis.999120.net ��������
�������Ƽ����� ������
�������Ƽ�������rhabdosarcomaofvulva
http://15910.dis.999120.net ��������
���������ܰ��� ������
���������ܰ����������νܽ������νܽ��ϲ���������������������Ƥϸ���������ܰ�������������״ϸ��������Hokdkin'sdiseaseofvulva
http://15911.dis.999120.net ��������
���������ϲ����� ������
���������ϲ�����
http://15912.dis.999120.net ��������
����¡��Ƥ����ά���� ������
����¡��Ƥ����ά����
http://15913.dis.999120.net ��������
������Ƥ������ ������
������Ƥ��������sarcomaepithelioidesofvulva
http://15914.dis.999120.net ��������
����������ά��֯ϸ���� ������
����������ά��֯ϸ������fibroxanthosarcoma����������ά��֯ϸ��������ά��ɫ����
http://15915.dis.999120.net ��������
���������� ������
������ά�����������Һ��ά������fibromasarcomatosumofvulva
http://15916.dis.999120.net ��������
����֬������ ������
����֬��������adiposesarcomaofvulva��lipoblastomaofvulva��������֬ϸ����������֬ĸϸ����
http://15917.dis.999120.net ��������
�������Կ���ϸ���� ������
�������Կ���ϸ�������������Կ���ϸ��ʩ��ϸ����
http://15918.dis.999120.net ��������
�������Ժ��Ƽ����� ������
�������Ժ��Ƽ�����
http://15919.dis.999120.net ��������
������״��Ƥ�� ������
������״��Ƥ����pavement-epitheliumcarcinomaofvagina��tabularepitheliumcarcinomaofvagina��������ƽ��Ƥ��
http://15920.dis.999120.net ��������
����ת�������� ������
����ת��������������ת����
http://15921.dis.999120.net ��������
������ɫ���� ������
������ɫ����������������
http://15922.dis.999120.net ��������
����������� ������
������������������ѻ��������������߲����
http://15923.dis.999120.net ��������
������Ϣ�� ������
������Ϣ�⣻polypusofvaginalwall������Ϣ��
http://15924.dis.999120.net ��������
�����ٲ� ������
�����ٲ�
http://15925.dis.999120.net ��������
�����ٰ� ������
�����ٰ���adenocarcinomavagina
http://15926.dis.999120.net ��������
�������Ƽ����� ������
�������Ƽ�������rhabdosarcomaofvagina
http://15927.dis.999120.net ��������
����ƽ�������� ������
����ƽ����������liomyosarcomaofvagina
http://15928.dis.999120.net ��������
�����������չܻ���� ������
�����������չܻ�����������������չܻ����
http://15929.dis.999120.net ��������
����Ѫ������ ������
����Ѫ��������hemangiosarcomaofvagina
http://15930.dis.999120.net ��������
�����ӹ���Ĥ�������� ������
�����ӹ���Ĥ��������
http://15931.dis.999120.net ��������
�ӹ���ƽ������ ������
�ӹ���ƽ��������fibroidofcervix��liomyomaofcervix��myomalevicellulareofcervix
http://15934.dis.999120.net ��������
������Ƥ������ ������
������Ƥ�����䣻������Ƥ������cervicalintraepithelialneoplasia
http://15935.dis.999120.net ��������
����С�ͽ��� ������
����С�ͽ����������½���������������ԭλ����С������������ڼ��ʽ����������ڽ����������ͽ����������ͽ���
http://15936.dis.999120.net ��������
�������� ������
��������invasivecarcinomaofcervix���ӹ�����������
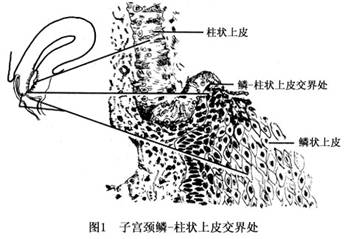
http://15937.dis.999120.net ��������
�����ٰ� ������
�����ٰ���adenocarcinomaoftheuterinecervix
http://15938.dis.999120.net ��������
���������� ������
����������
http://15939.dis.999120.net ��������
�ӹ����ж˰� ������
�ӹ����ж˰���carcinomaofcervicalstump
http://15940.dis.999120.net ��������
����ϲ������� ������
����ϲ���������pregnancycomplicatingcancerofthecervix
http://15942.dis.999120.net ��������
�ӹ����� ������
�ӹ�������hysteromyoma��leiomyomaofuterus��leiomyomauteri���ӹ�ƽ������
http://15943.dis.999120.net ��������
���ѹܰ�����֢ ������
���ѹܰ�����֢
http://15944.dis.999120.net ��������
�ӹ���Ĥ���� ������
�ӹ���Ĥ������hyperplasiaendometrii
http://15945.dis.999120.net ��������
�ӹ���ĤϢ�� ������
�ӹ���ĤϢ�⣻metropolypus���ӹ�Ϣ��
http://15946.dis.999120.net ��������
�ӹ���Ĥ�������� ������
�ӹ���Ĥ����������uterinestromalendometriosis��uterinestromatosis���ӹ��ܰ��ڼ��ʼ������ӹ��ܰ��ڼ�����λ֢���ӹ���Ĥ������λ֢
http://15948.dis.999120.net ��������
�ӹ���������Ҷ����� ������
�ӹ���������Ҷ�������malignantmullerianmixedtumor���ӹ����������ӹ��������չܻ�������ӹ����Ի��������Ҷ��
http://15949.dis.999120.net ��������
�ӹ���Ĥ�� ������
�ӹ���Ĥ����carcinomaofuterinecorpus��endomerioidadenocarcinoma���ӹ���Ĥ�ٰ����ӹ���Ĥ���ٰ����ӹ��尩��adenocarcinomaendometrium��endometrialcancer
http://15950.dis.999120.net ��������
ԭ�������ѹܰ� ������
ԭ�������ѹܰ���ԭ���Է�¦Ƥŷ�ܰ���ԭ���Է�¦Ƥŷ�ϰ�
http://15951.dis.999120.net ��������
����ϲ��ӹ����� ������
����ϲ��ӹ�������pregnancyassociatedwithleiomyomauteri��pregnancyassociatedwithuterusmyoma
http://15952.dis.999120.net ��������
�ѳ���Һ������ ������
�ѳ���Һ������
http://15953.dis.999120.net ��������
�ѳ��Һ������ ������
�ѳ��Һ���������ѳ�ճҺ�������ѳ�ճҺ������
http://15954.dis.999120.net ��������
�ѳ���ϸ������ ������
�ѳ���ϸ��������celluleclearofovary��wasserhellecellsofovary��water-clearcellofovary���ѳ���ϸ������
http://15956.dis.999120.net ��������
�ѳ����������� ������
�ѳ�����������
http://15957.dis.999120.net ��������
�ѳ�����ϸ���� ������
�ѳ�����ϸ������ovariangranulosacelltumors
http://15958.dis.999120.net ��������
�ѳ���Ĥϸ���� ������
�ѳ���Ĥϸ������ovarianthecacelltumors��thecomaofovary
http://15959.dis.999120.net ��������
����ۺ��� ������
����ۺ�����Demons-Meigs�ۺ�����Domons-Meigs�����ۺ�����Meigs-Cass�ۺ�����Meigs-Salmon�ۺ������ѳ���-��ˮ-��ˮ�ۺ������ѳ���-�ظ�ǻ��Һ�ۺ������ѳ�-��ˮ-��ˮ�ۺ������ѳ����ظ�ǻ��Һ�ۺ���������ۺ�֢��÷��˹���ۺ�����÷��˹�ۺ�����÷-�������ۺ�������ǻ���ϲ���-��ˮ��
http://15961.dis.999120.net ��������
�ѳ�Ӳ���Լ����� ������
�ѳ�Ӳ���Լ�����
http://15963.dis.999120.net ��������
�ѳ�֧�ּ���ϸ���� ������
�ѳ�֧�ּ���ϸ����
http://15964.dis.999120.net ��������
�ѳ�����״�������� ������
�ѳ�����״�����������ѳ��������黷״С��
http://15965.dis.999120.net ��������
�ѳ�����ĸϸ���� ������
�ѳ�����ĸϸ�������ѳ�������ϸ����
http://15966.dis.999120.net ��������
�ѳ������̥�� ������
�ѳ������̥����benignovarianteratomaofovary���ѳ���Ƥ�����ף��ѳ������ͻ�̥�����ѳ������Ի�̥�����ѳ����Ի�̥�����ѳ����Ի�̥��

http://15967.dis.999120.net ��������
�ѳ�δ�����̥�� ������
�ѳ�δ�����̥�����ѳ��dz����ͻ�̥�����ѳ�δ�����Ի�̥�����ѳ������ͻ�̥��
http://15968.dis.999120.net ��������
�ѳ��ѻ����� ������
�ѳ��ѻ��������ѳ��������
http://15969.dis.999120.net ��������
ԭ�����ѳ��ް� ������
ԭ�����ѳ��ް���non-gestationalovalovarianchoriocarcinoma���ѳ����������ް���ԭ�����ѳ���ëĤ��
http://15971.dis.999120.net ��������
�ѳ��������ֳϸ��-������������ ������
�ѳ��������ֳϸ��-������������
http://15972.dis.999120.net ��������
θ������ת���ѳ� ������
θ������ת���ѳ����ѳ��̷���θ������
http://15973.dis.999120.net ��������
�ѳ���ά��֯��Դ���� ������
�ѳ���ά��֯��Դ����
http://15974.dis.999120.net ��������
�������� ������
�������ף�luteincyst
http://15975.dis.999120.net ��������
�����ѳ��ۺ�֢ ������
�����ѳ��ۺ�����polycysticovarysyndrome��Rokitandky����Stein-Leventhal�ۺ����������ѳ��ۺ�֢���������ѳ��ۺ�����˫������ѳ��ۺ�֢
http://15976.dis.999120.net ��������
������ϸ����ֳ�ۺ��� ������
������ϸ����ֳ�ۺ���������Ĥϸ����ֳ�ۺ�����������ϸ����ֳ�ۺ�֢
http://15977.dis.999120.net ��������
�ѳ������� ������
�ѳ�������
http://15978.dis.999120.net ��������
�������ѳ��������� ������
�������ѳ������������������ѳ�������
http://15980.dis.999120.net ��������
�Ŵ������ٰ�-�ѳ����ۺ��� ������
�Ŵ������ٰ�-�ѳ����ۺ��������ٰ�-�ѳ����ۺ������Ŵ������ٰ�-�ѳ������Ŵ������ٰ�-�ѳ����ۺ�֢
http://15981.dis.999120.net ��������
ԭ�����ѳ���ۺ��� ������
ԭ�����ѳ���ۺ�����ԭ�����ѳ���ۺ�֢��ԭ�����ѳ���-ɣ�����ۺ���
http://15982.dis.999120.net ��������
�ѳ������ۺ��� ������
�ѳ������ۺ������ѳ������ۺ�֢��ʣ���ѳ��ۺ���
http://15983.dis.999120.net ��������
�ѳ�������Դ���� ������
�ѳ�������Դ����
http://15984.dis.999120.net ��������
�ѳ����Ը�Ĥ��Ƥ�� ������
�ѳ����Ը�Ĥ��Ƥ�����ѳ���Ĥ����
http://15985.dis.999120.net ��������
�ѳ�Сϸ���� ������
�ѳ�Сϸ������ovarianoatcellcarcinoma��ovariansmallcellcancer��ovariansmall-cellcarcinoma���ѳ�����ϸ�������ѳ�������ϸ����
http://15986.dis.999120.net ��������
�ѳ���������Ҷ����� ������
�ѳ���������Ҷ��������ѳ����������ѳ����Ի��������Ҷ�����ѳ����չܻ����
http://15987.dis.999120.net ��������
Ů����ֳ���ಿλԭ���� ������
Ů����ֳ���ಿλԭ����
http://15988.dis.999120.net ��������
�ѳ��ض�ˮ�� ������
�ѳ��ض�ˮ�ף�massiveedemaoftheovary���ѳ���Ƭˮ��
http://15989.dis.999120.net ��������
�ѳ���ά���� ������
�ѳ���ά�������ѳ�������ά��֯����
http://15990.dis.999120.net ��������
�������� ������
�������ף�solitaryfolliclecyst���������������ף��������ף�ë������
http://15991.dis.999120.net ��������
����ϸ������ ������
����ϸ��������������ϸ����������������������ϸ������������Ҷ������trophoblasticdisease�����������ȵ�ϸ������������ϸ������

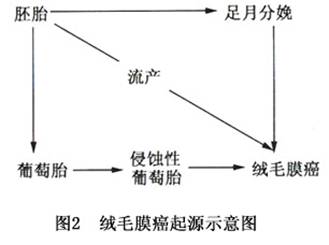
http://15992.dis.999120.net ��������
�����꼰С��������ϸ���� ������
�����꼰С��������ϸ����
http://15994.dis.999120.net ��������
�����꼰С���ӹ����� ������
�����꼰С���ӹ�����
http://15995.dis.999120.net ��������
�����꼰С���ѳ����� ������
�����꼰С���ѳ�����

http://15996.dis.999120.net ��������
����Ӳ��ά�� ������
����Ӳ��ά�������ڳ���ά�������ڴ�״�������ڸ�������ά������������Ϯ����ά�����������ʹ�����ά����������ά������������ά��֯��������
http://15997.dis.999120.net ��������
������������������ ������
�����������������ѣ�rupturedabdominalaneurysm�������Ը�����������rupturedabdominalaorticaneurysm
http://15998.dis.999120.net ��������
��֢�Ը��������� ������
��֢�Ը��������������Ը���������
http://15999.dis.999120.net ��������
��Ⱦ�Ը��������� ������
��Ⱦ�Ը���������
http://16000.dis.999120.net ��������
�ϲ��������ĸ��������� ������
�ϲ��������ĸ������������ϲ����������ĸ���������
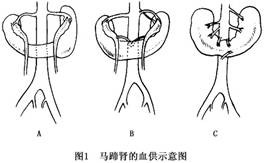
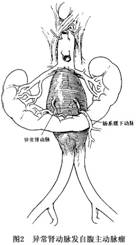
http://16001.dis.999120.net ��������
���������� ������
������������abdominalaneurysm
http://16002.dis.999120.net ��������
���������� ������
������������fibromasarcomatosumofabdominalwall
http://16003.dis.999120.net ��������
��Ĥ��Ƥ�� ������
��Ĥ��Ƥ����mesotheliomaofpleura����Ĥ��Ƥϸ����
http://16004.dis.999120.net ��������
��Ĥ���Һ�� ������
��Ĥ���Һ������Ĥ����ճҺ��
http://16005.dis.999120.net ��������
ԭ���Ը�Ĥ�� ������
ԭ���Ը�Ĥ����ԭ���Ը�Ĥ��ͷ��Һ��
http://16006.dis.999120.net ��������
��Ĥת�ư� ������
��Ĥת�ư���ת���Ը�Ĥ����
http://16007.dis.999120.net ��������
��Ĥ��֬���� ������
��Ĥ��֬������retroperitonealliparomphalus��retroperitoneallipoma��ԭ���Ը�Ĥ��֬����
http://16008.dis.999120.net ��������
ԭ���Ը�Ĥ������ ������
ԭ���Ը�Ĥ������
http://16009.dis.999120.net ��������
�����Ը�Ĥ������ ������
�����Ը�Ĥ��������recurrentretroperitonealtumour���ٷ��Ը�Ĥ������
http://16010.dis.999120.net ��������
ԭ���Գ�ϵĤ���� ������
ԭ���Գ�ϵĤ����
http://16012.dis.999120.net ��������
��ϵĤ������ ������
��ϵĤ������
http://16013.dis.999120.net ��������
��ϵĤ���� ������
��ϵĤ����
http://16014.dis.999120.net ��������
����Ѫ���� ������
����Ѫ����
http://16015.dis.999120.net ��������
����� ������
�����
http://16016.dis.999120.net ��������
�κ���״Ѫ���� ������
�κ���״Ѫ����
http://16017.dis.999120.net ��������
������ ������
��������hepaticadenoma��hepatocellularadenoma����ϸ����������������
http://16018.dis.999120.net ��������
����� ������
�δ�������mesenchymalhamartomaofliver���μ�Ҷ�Դ����������������
http://16019.dis.999120.net ��������
��Ѫ��ƽ����֬���� ������
��Ѫ��ƽ����֬������angioleiomyolipomaofliver����Ѫ�ܼ�֬��
http://16020.dis.999120.net ��������
ԭ���Ըΰ� ������
ԭ���Ըΰ����ΰ�
http://16021.dis.999120.net ��������
����� ������
�����
http://16022.dis.999120.net ��������
����ά��㰩 ������
����ά��㰩
http://16023.dis.999120.net ��������
ԭ���Ը�֬������ ������
ԭ���Ը�֬��������ԭ���Ը�֬����
http://16024.dis.999120.net ��������
ת���Ըΰ� ������
ת���Ըΰ���secondarylivercarcinoma���̷��Ըΰ���metastaticlivercancer������ת����
http://16025.dis.999120.net ��������
��ĸϸ���� ������
��ĸϸ����������̥��������ϸ������Ӥ����ϸ�������ζ�ϸ����
http://16026.dis.999120.net ��������
��Ѫ������ ������
��Ѫ���������ζ���Ѫ����Ƥ�����ο��ո�ϸ������������״��Ƥϸ��������Ѫ����Ƥϸ��������hepaticangiosarcoma��Kupfferϸ��������malignanthemangiomaofliver����Ѫ����Ƥ�������������Ѫ����������Ѫ���������⸥ϸ������
http://16027.dis.999120.net ��������
����Ƥ��Ѫ����Ƥϸ���� ������
����Ƥ��Ѫ����Ƥϸ����������Ƥ��Ѫ����Ƥ��
http://16028.dis.999120.net ��������
Ϣ�� ������
��������������benigntumorofgallbladder������¡���Բ��䣻������ͷ״����Ϣ��

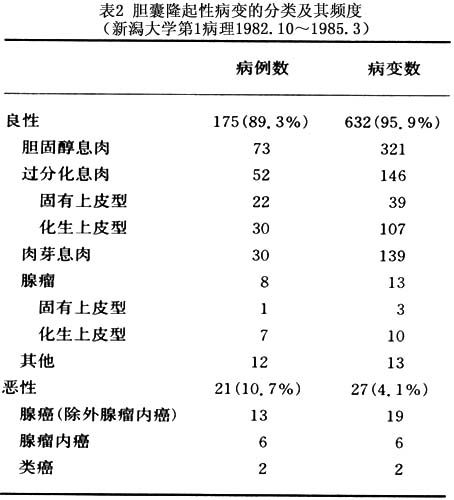
http://16029.dis.999120.net ��������
���Ұ� ������
���Ұ�
http://16030.dis.999120.net ��������
����ƽ�������� ������
����ƽ����������leiomyosarcomaofgallbladder
http://16031.dis.999120.net ��������
����� ������
�����
http://16032.dis.999120.net ��������
�������� ������
������Χ����periampullarycarcinoma�����غ�����
http://16033.dis.999120.net ��������
���ܰ� ������
���ܰ���cholangiocarcinoma������ϸ����
http://16034.dis.999120.net ��������
���ٰ� ������
���ٰ���cancerofpancreas��pancreaticcarcinoma
http://16035.dis.999120.net ��������
� ������
��ۺ�������-ɣ�����ۺ�������-ɣ�ۺ�������Ƥ�����ಡ���������ۺ�������ۺ�֢����ԭ���������ϸ����������ϸ������argentaffinoma
http://16036.dis.999120.net ��������
�ȵ����� ������
�ȵ�������insuloma���ȵ�Bϸ�������ȵ���ϸ�������ȵ���
http://16037.dis.999120.net ��������
�ȸ�Ѫ������ ������
�ȸ�Ѫ������
http://16038.dis.999120.net ��������
�ȶ����� ������
�ȶ�������pancreaticpolypeptide-producingtumor
http://16039.dis.999120.net ��������
�������� ������
����������adenosarcomaofpancreas��sarcoadenomaofpancreas
http://16040.dis.999120.net ��������
Ƣ�������� ������
Ƣ��������
http://16042.dis.999120.net ��������
Ƣ������ ������
Ƣ������
http://16043.dis.999120.net ��������
Ƣ��ת�������� ������
Ƣ��ת����������Ƣ��ת����
http://16045.dis.999120.net ��������
ʮ��ָ���������� ������
ʮ��ָ������������benigntumordodecadactylon��benigntumorzwolffingerdarm
http://16046.dis.999120.net ��������
ʮ��ָ��� ������
ʮ��ָ�����carcinoidofdodecadactylon��carcinoidofzwolffingerdarm��ʮ��ָ�����
http://16047.dis.999120.net ��������
ʮ��ָ���ٰ� ������
ʮ��ָ���ٰ���adenocarcinomaofdodecadactylon��adenocarcinomaofzwolffingerdarm
http://16048.dis.999120.net ��������
ʮ��ָ��ƽ�������� ������
ʮ��ָ��ƽ����������leiomyosarcomaofdodecadactylo��leiomyosarcomaofdodecadactylon��leiomyosarcomaofzwolffingerdarm��liomyosarcomaofduodenum
http://16049.dis.999120.net ��������
������ ������
������
http://16051.dis.999120.net ��������
��Ϣ�� ������
С��������intestinadadenoma����Ϣ��
http://16052.dis.999120.net ��������
С��ƽ������ ������
С��ƽ��������liomyomaofsmallintestine��myomalevicellulareofsmallintestine
http://16053.dis.999120.net ��������
С��֬���� ������
С��֬������liparomphalusofsmallintestine��pimelomaofsmallintestine
http://16054.dis.999120.net ��������
С��Ѫ���� ������
С��Ѫ������intestinalangioma��angeiomaofsmallintestine��angioneoplasmofsmallintestine��haemangiomaofsmallintestine��hemangiomaofsmallintestine��vasculartumorofsmallintestine
http://16055.dis.999120.net ��������
С����ά�� ������
С����ά����fibroidtumorofsmallintestine��inomaofsmallintestine
http://16056.dis.999120.net ��������
С��ԭ���Զ����ܰ��� ������
С��ԭ���Զ����ܰ�����primarysmallintestinallymphoma��primarysmallintestinalmalignantlymphoma
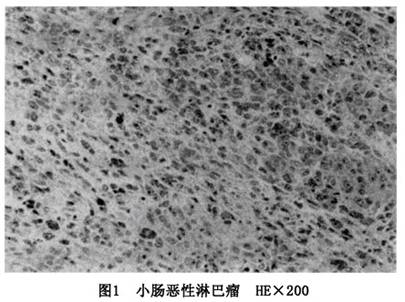
http://16057.dis.999120.net ��������
С���ٰ� ������
С���ٰ���intestinaladenocarcinoma��glandsofsmallintestine��ϵĤС����
http://16058.dis.999120.net ��������
С��ƽ�������� ������
С��ƽ����������intestinalleiomyosarcoma��liomyosarcomaofsmallintestine
http://16059.dis.999120.net ��������
��� ������
С�����entericcarcinoid��APUDϸ������С������ϸ������С�����ڷ���������С������ϸ����
http://16060.dis.999120.net ��������
ת����С������ ������
ת����С��������С��ת����
http://16061.dis.999120.net ��������
��ֱ��Ѫ���� ������
��ֱ��Ѫ������angeiomaofthecolonandrectum��angiomaofthecolonandrectum��angioneoplasmofthecolonandrectum��hemangiomaofthecolonandrectum��hemartomaofthecolonandrectum��vasculartumorofthecolonandrectum
http://16062.dis.999120.net ��������
�᳦ƽ������ ������
�᳦ƽ��������fibroidofcolon��liomyomaofcolon��myomalevicellulareofcolon
http://16063.dis.999120.net ��������
�᳦�� ������
�᳦����coloniccarcinoma���᳦��֯���Բ���
http://16064.dis.999120.net ��������
�᳦� ������
�᳦����᳦�ȸ�ϸ�������᳦����ϸ�������᳦���������ڷ�������
http://16065.dis.999120.net ��������
��֬���� ������
��֬������liparomphalusoflargeintestine��pimelomaoflargeintestine
http://16067.dis.999120.net ��������
��Ѫ���� ������
��Ѫ����
http://16068.dis.999120.net ��������
�� ������
��
http://16069.dis.999120.net ��������
��ƽ�������� ������
��ƽ��������
http://16070.dis.999120.net ��������
�عܰ� ������
�عܰ���cancerofanalcanal���عܶ�������
http://16072.dis.999120.net ��������
�ع�ֱ�����Ժ�ɫ���� ������
�ع�ֱ�����Ժ�ɫ�������ع�ֱ�����Ժ�����������ֱ�����Ժ�ɫ����������ֱ�����Ժ�����
http://16073.dis.999120.net ��������
�������� ������
������������������������������
http://16074.dis.999120.net ��������
��β� ������
��β�
http://16075.dis.999120.net ��������
��β�ٰ� ������
��β�ٰ���adenocarcinomaofappendix
http://16076.dis.999120.net ��������
����� ������
�������tumorofappendix
http://16077.dis.999120.net ��������
�������� ������
����������renalaneurysm
http://16078.dis.999120.net ��������
���۰� ������
���۰���renalpelviccarcinoma����������
http://16079.dis.999120.net ��������
���� ������
��ϸ������adenocarcinomaofkidney��dear-cellcarcinomaofkidney����������ʵ�ʰ������ٰ�����ϸ������hypernephroidcarcinoma��renalcellcarcinomaofkidney������������
http://16080.dis.999120.net ��������
������ϸ���� ������
������ϸ������oncocytomarenalcellcarcinoma����������ϸ����
http://16081.dis.999120.net ��������
��Թ����������� ������
��Թ�������������myelomacastnephropathy������������������lightchainnephropathy�������ز��������������ϲ���������������
http://16082.dis.999120.net ��������
ʵ������������ ������
ʵ������������ʵ��������������
http://16083.dis.999120.net ��������
�ܰ������������ ������
�ܰ���������������ܰ���
http://16084.dis.999120.net ��������
��ĸϸ���� ������
��ĸϸ������embryonalcarcinoscarcome��Wilmstumor��Wilms�����������������������������̥��������̥�Զ��Ի����������ϸ������ά��ķ˹����embryomaofkidney��Whilmstumor��Wilmtumorofkidney������̥ĸϸ����������̥��������Wilm��stumor
http://16085.dis.999120.net ��������
�������� ������
��������
http://16086.dis.999120.net ��������
�ȸ�ϸ���� ������
�ȸ�ϸ������chromaffintumor��chromaffin-celltumor��chromaffinoma��chromophiletumor��medullarychromaffinoma��medullosuprarenoma�������������������������ȸ�ĸϸ������phaeochromocytoma
http://16087.dis.999120.net ��������
������������ ������
��������������������ż����
http://16088.dis.999120.net ��������
��������� ������
���������
http://16089.dis.999120.net ��������
������� ������
�������
http://16090.dis.999120.net ��������
��������� ������
���������
http://16091.dis.999120.net ��������
�ǰ� ������
������osseoustumor��osteocarcinoma��osteoncus���ǰ�
http://16092.dis.999120.net ��������
�������� ������
����������osteoidosteoma
http://16093.dis.999120.net ��������
������������� ������
���������������multiplehereditarydeformingchondrody��Ollier��������Ƿ����쳣�������������������������Ƿ����쳣������Ŵ����������Ƿ����쳣����Ŵ����������Ƿ����쳣������������������
http://16096.dis.999120.net ��������
��Ĥ������ ������
��Ĥ����������Ƥ������������ƫ����������
http://16097.dis.999120.net ��������
�����Թ������� ������
�����Թ������������¹��ࣻ��������
http://16098.dis.999120.net ��������
�Ŵ��Զ�Թ������� ������
�Ŵ��Զ�Թ�������������������ࣻ�Ǹɶ�����֢���Ŵ��Ի��������Ƿ����쳣֢
http://16099.dis.999120.net ��������
����ĸϸ���� ������
����ĸϸ������Codman������������ϸ�������ƻ���ϸ���������������Ծ�ϸ�����������Ǿ�ϸ���������Թ�������ĸϸ����
http://16100.dis.999120.net ��������
�����Һ����ά�� ������
�����Һ����ά��������ճҺ����ά������ά�Һ��������
http://16101.dis.999120.net ��������
�Ǿ�ϸ���� ������
�Ǿ�ϸ������osteoclastoma���ƹ�ϸ����
http://16102.dis.999120.net ��������
���ܰ��� ������
���ܰ�����angiolymphomaofbone��angiomalymphaticumofbone�����ܰ�����
http://16103.dis.999120.net ��������
��Ѫ���� ������
��Ѫ������osteoangioma
http://16104.dis.999120.net ��������
�ɶ����� ������
�ɶ�����
http://16105.dis.999120.net ��������
�N������ ������
�N������
http://16106.dis.999120.net ��������
��������ά��֯ϸ���� ������
��������ά��֯ϸ������benignfibrohistiocytomaofbone
http://16107.dis.999120.net ��������
�������� ������
��������
http://16108.dis.999120.net ��������
�������� ������
����������fibroneuromaofbone��neuroinomaofbone
http://16109.dis.999120.net ��������
�������������� ������
�������������ף�����������������
http://16110.dis.999120.net ��������
����ά�ṹ���� ������
����ά������ֳ֢��fibrousdysplasiaofbone��fibroushyperplasiaofbone������ά�ṹ��������ά�Թǽṹ����
http://16111.dis.999120.net ��������
��֯ϸ����ά�� ������
��֯ϸ����ά����fibrohistiocytoma���dzɹ�����ά�����ǹǻ�����ά���������˴���������������ά��ȱ����άƤ��ȱ����ά��֯ϸ����
http://16112.dis.999120.net ��������
��Ѫ����Ƥϸ���� ������
��Ѫ����Ƥϸ��������Ѫ����Ƥ��
http://16113.dis.999120.net ��������
��Ѫ����Ƥϸ���� ������
��Ѫ����Ƥϸ������haemangiopericytomaofbone��peritheliomaofbone������Ƥ��
http://16114.dis.999120.net ��������
��������ϸ���� ������
��������ϸ���������ǵij���ϸ��������������
http://16115.dis.999120.net ��������
������ ������
��������osteogenicsarcoma��osteomasarcomatosum���ɹ���������Դ��������ԭ����������ϸ������

http://16116.dis.999120.net ��������
ת���Թ����� ������
ת���Թ��������ǵ�ת��������ת���Զ�����������ת����
http://16117.dis.999120.net ��������
�������� ������
����������chondromasarcomatosum��sarcoenchondroma
http://16118.dis.999120.net ��������
�������� ������
����������δ�ֻ���֯ϸ����������������
http://16119.dis.999120.net ��������
�Ƕ����ܰ��� ������
�Ƕ����ܰ�������֯ϸ�������νܽ�
http://16120.dis.999120.net ��������
ԭ���Թ��ܰ��� ������
ԭ���Թ��ܰ�����ԭ���Թ���״ϸ��������primarylymphomaofbone
http://16121.dis.999120.net ��������
��Ѫ������ ������
��Ѫ��������hemangiosarcomaofbone�����Թ�Ѫ�������Ƕ���Ѫ����Ƥ��������������
http://16122.dis.999120.net ��������
�������� ������
����������osteofibrosarcoma
http://16123.dis.999120.net ��������
�Ƕ�����ά��֯ϸ���� ������
�Ƕ�����ά��֯ϸ������malignantfibroushistiocytomaofbone���Ƕ���ά��֯ϸ����
http://16124.dis.999120.net ��������
��Ĥ���� ������
��Ĥ������malignantsynovioma��synovialsarcoma�����Ի�Ĥ��
http://16125.dis.999120.net ��������
���Ƽ����� ������
���Ƽ�������rhabdosarcoma
http://16126.dis.999120.net ��������
����������ѿ�� ������
����������ѿ�ף��������ϸ����ѿ�ף�������ϸ����ѿ�ף�������ϸ������ѿ��
http://16127.dis.999120.net ��������
�����Թǻ��Լ��� ������
�����Թǻ��Լ��ף�myositisossificanstraumatica�������Թǻ����ǻ���Ѫ�ף���������Է������Թ������γɣ����ԹDz��������Թǻ��Լ��ף���������λ�ǻ�
http://16128.dis.999120.net ��������
�����Թؽ��� ������
�����Թؽ���
http://16129.dis.999120.net ��������
������������ƶѪ ������
������������ƶѪ������������ƶѪ
http://16130.dis.999120.net ��������
ԭ����������ϵͳ�ܰ��� ������
ԭ����������ϵͳ�ܰ�������״ϸ��������С����ϸ������Ѫ��������
http://16131.dis.999120.net ��������
غ���ܰ��� ������
غ���ܰ�����testiclymphadenoma��غ���ܰ�����
http://16132.dis.999120.net ��������
ԭ���Ա�ǻ�ܰ��� ������
ԭ���Ա�ǻ�ܰ���
http://16133.dis.999120.net ��������
ԭ���������ܰ��� ������
ԭ���������ܰ���
http://16135.dis.999120.net ��������
ԭ����Ƥ��Bϸ���ܰ��� ������
ԭ����Ƥ��Bϸ���ܰ�����primarycutaneousB-celllymphoma
http://16136.dis.999120.net ��������
ԭ�����������ܰ��� ������
ԭ�����������ܰ�����lymphomaprimaryeffusion��primaryeffusionlymphoma
http://16137.dis.999120.net ��������
Ƥ��֬Ĥ����Tϸ���ܰ��� ������
Ƥ��֬Ĥ����Tϸ���ܰ�����Ƥ��֬Ĥ����T-ϸ���ܰ�����subcutaneouspanniculitictcelllymphoma
http://16138.dis.999120.net ��������
���������ȱ���ۺ�������ܰ��� ������
���������ȱ���ۺ�������ܰ��������̲�����ܰ��������������ȱ���ۺ�������ܰ���
http://16139.dis.999120.net ��������
���������쳣�ۺ��� ������
���������쳣�ۺ��������������쳣�ۺ�֢
http://16141.dis.999120.net ��������
������֯ϸ���� ������
������֯ϸ������Tϸ���ܰ�����������״ϸ������������֯ϸ������֢�����飻�ǰ�Ѫ����״��Ƥϸ������֢���ܰ���״����������״ϸ����Ѫ������֯ϸ����������״ϸ������֢
http://16142.dis.999120.net ��������
�����ܰ�ϸ����Ѫ�� ������
�����ܰ�ϸ����Ѫ�������ܣ����Գ��ܰ�ϸ����Ѫ����������Ѫ��
http://16143.dis.999120.net ��������
�ʴ�ϸ����Ѫ�� ������
�ʴ�ϸ����Ѫ������֯�ȼ�ϸ����Ѫ��
http://16144.dis.999120.net ��������
��ϸ����Ѫ�� ������
��ϸ����Ѫ������ϸ����Ѫ����Plasmacyticleukemia
http://16145.dis.999120.net ��������
��������ϸ����Ѫ�� ������
��������ϸ����Ѫ��������ϸ����Ѫ��
http://16146.dis.999120.net ��������
�ȼ�����ϸ����Ѫ�� ������
�ȼ�����ϸ����Ѫ�����ȼ�ϸ����Ѫ����basophiliccellleukemia
http://16147.dis.999120.net ��������
������Ѫ�� ������
������Ѫ����congenitalleukemi��ԭ����Ѫ��
http://16148.dis.999120.net ��������
�� ������
������ϵͳ��Ѫ������
http://16149.dis.999120.net ��������
�̷���Ѫ�� ������
�̷���Ѫ����t-MDS/AML��������ع��������쳣�ۺ���/������ϵ��Ѫ��
http://16150.dis.999120.net ��������
�ܰ���ϸ����Ѫ�� ������
�ܰ���ϸ����Ѫ����LSL��lymphosarcomacellleukemia���ܰ�����ϸ����Ѫ��
http://16151.dis.999120.net ��������
����Tϸ����Ѫ�� ������
����Tϸ����Ѫ��������Tϸ����Ѫ����������Tϸ����Ѫ����adultT-cellleukemia/lymphoma������Tϸ����Ѫ��/�ܰ���
http://16152.dis.999120.net ��������
�������Լ���Ѫ�� ������
�������Լ���Ѫ������Ѫ��ǰ�ڣ�ð����Ѫ��
http://16153.dis.999120.net ��������
������ϸ����Ѫ�� ������
������ϸ����Ѫ����ANLL�������ܣ����������Է��ܰ�ϸ����Ѫ����������ϸ����Ѫ��
http://16154.dis.999120.net ��������
����ϲ���Ѫ�� ������
����ϲ���Ѫ����pregnancycomplicatingleucocythemia��pregnancycomplicatingleukaemia��pregnancycomplicatingleukecythemia��pregnancycomplicatingleukocythemia������ϲ���Ѫ�����֢
http://16155.dis.999120.net ��������
��Ѫ���鷢�ľ����ϰ� ������
��Ѫ���鷢�ľ����ϰ�����Ѫ���鷢�ľ�����Ѫ���鷢�ľ�����ң���Ѫ���鷢�ľ�������
http://16156.dis.999120.net ��������
�����Ӻ���Ѫ�� ������
�����Ӻ���Ѫ����acutemixedleukemia�����Ի��ϸ����Ѫ�����ӽ��Լ���Ѫ��
http://16157.dis.999120.net ��������
����������ϸ����Ѫ�� ������
����������ϸ����Ѫ��������ǰ��ϸ����Ѫ��������������ϸ����Ѫ��
http://16158.dis.999120.net ��������
���ܰ�ϸ����Ѫ�� ������
���ܰ�ϸ����Ѫ����ǰ�ܰ�ϸ����Ѫ�������ܰ�ϸ����Ѫ��
http://16159.dis.999120.net ��������
ëϸ����Ѫ�� ������
ëϸ����Ѫ����ëϸ����Ѫ������Ѫ������״��Ƥϸ����������ëϸ����Ѫ����hairy-cellleukemia
http://16160.dis.999120.net ��������
������ϸ����Ѫ�� ������
������ϸ����Ѫ����������������ϸ����Ѫ����������ϸ����Ѫ����������ϸ����Ѫ��
http://16161.dis.999120.net ��������
�����ܰ�ϸ����Ѫ�� ������
�����ܰ�ϸ����Ѫ�������ܣ������ܰ�ϸ����Ѫ��
http://16162.dis.999120.net ��������
������ܰ�ϸ����Ѫ�� ������
������ܰ�ϸ����Ѫ����T���ܰ�ϸ����ֳ�Լ�����T�����ܰ�ϸ����Ѫ����������ܰ�ϸ�����ܰ�ϸ����ֳ�Լ�����������ܰ�ϸ����Ѫ��
http://16163.dis.999120.net ��������
����ϲ��ܰ��� ������
����ϲ��ܰ�����pregnancycomplicatingleucoma��pregnancycomplicatingleukoma��pregnancycomplicatinglymphadenoma������ϲ��ܰ�����
http://16164.dis.999120.net ��������
�ܰ�������ѿ�� ������
�ܰ�������ѿ�ף��ܰ�������ѿ�ײ���polymorphicreticulosis����������״ϸ������֢������Ѫ������ѿ�ײ���Ѫ���������ܰ�����Ѫ��������������ֳ�Բ��䣻������״ϸ������
http://16165.dis.999120.net ��������
����� ������
������νܽ�������ܰͲ����ܰ���ѿ�ף��ܰ���״ϸ��������-˹����Hodgkin'sdisease���νܽ��ϲ�
http://16166.dis.999120.net ��������
�ǻ�����ܰ��� ������
�ǻ�����ܰ������Ǻνܽ��ܰ������ǻ��������ܰ�����
http://16167.dis.999120.net ��������
�Ĥ�������֯�ܰ��� ������
�Ĥ�������֯�ܰ�����ճĤ�������֯�ܰ������Ĥ�������֯�ܰ�������ճĤ�������֯�ܰ�����
http://16168.dis.999120.net ��������
ԭ���������ܰ��� ������
ԭ���������ܰ���
http://16169.dis.999120.net ��������
���ٰ�Ƥ��ת�� ������
���ٰ�Ƥ��ת�ƣ�����
http://16170.dis.999120.net ��������
�ΰ�Ƥ��ת�� ������
�ΰ�Ƥ��ת�ƣ��ΰ��̷���Ƥ��ת��
http://16171.dis.999120.net ��������
θ������Ƥ��ת�� ������
θ������Ƥ��ת�ƣ�Ƥ���̷���θ������
http://16172.dis.999120.net ��������
��ǻ��Ƥ��ת�� ������
��ǻ��Ƥ��ת�ƣ���ǻ���̷���Ƥ��ת��
http://16173.dis.999120.net ��������
���������Ƥ��ת�� ������
���������Ƥ��ת��
http://16174.dis.999120.net ��������
��ֳ������Ƥ��ת�� ������
��ֳ������Ƥ��ת�ƣ�genitalducttumors
http://16175.dis.999120.net ��������
���ڷ�������Ƥ��ת�� ������
���ڷ�������Ƥ��ת��
http://16176.dis.999120.net ��������
����ϸ��֬���� ������
����ϸ��֬����
http://16178.dis.999120.net ��������
ƽ�������� ������
ƽ����������liomyosarcoma
http://16179.dis.999120.net ��������
ƽ���������� ������
ƽ����������
http://16180.dis.999120.net ��������
Ƥ�������� ������
Ƥ����������cutaneouschondriom��cutaneouschondroma��cutaneouschondrophyma��cutaneousecchondrosisphysaliformis
http://16181.dis.999120.net ��������
Ƥ������ ������
Ƥ��������dermostosis��Ƥ���ǻ�
http://16182.dis.999120.net ��������
����֬��ĸϸ���� ������
����֬��ĸϸ������embryonallipoma������֬������������֬ĸϸ��������̥��֬���������Գ�֬ϸ����
http://16183.dis.999120.net ��������
����֬�������������Ƥ�� ������
����֬�������������Ƥ������֬�������������Ƥ��
http://16184.dis.999120.net ��������
��ɫ�� ������
��ɫ����fibromalipoidicum��fibromalipomatodes��xanthomatosis������������������֬��������֬����ά����֬������ά����֬������ά��������֢�����ಡ
http://16185.dis.999120.net ��������
֬���� ������
֬������pimeloma
http://16186.dis.999120.net ��������
������֬���� ������
������֬����
http://16187.dis.999120.net ��������
�ݷ�֬�� ������
�ݷ�֬����brownfattumor������������ɫ֬����
http://16188.dis.999120.net ��������
Ѫ��֬���� ������
Ѫ��֬������Ѫ��֬��
http://16189.dis.999120.net ��������
dz����Ƥ��֬������ ������
dz����Ƥ��֬�����룻nevuslipomatosussuperficialis��dz����֬�������룻dz����֬��������
http://16190.dis.999120.net ��������
֬������ ������
֬������֬��������lipomasarcomatodes��sarcolipoma
http://16191.dis.999120.net ��������
Ƥ��ƽ������ ������
Ƥ��ƽ��������cuteneousleiomyoma
http://16192.dis.999120.net ��������
Ƥ���ܰ�ϸ���� ������
Ƥ���ܰ�ϸ������BafverstedtƤ�������ܰͽᲡ��LymphadenosisBenignaCutisofBafverstedt��Spiegler-fendt�����ܰ�����Spiegler-fendt��������cutaneouslymphoma
http://16193.dis.999120.net ��������
��ϸ����Ƥ�� ������
��ϸ����Ƥ����Degos��Ƥ����palecellAcanthoma����ϸ����Ƥ������ϸ����Ƥ��
http://16194.dis.999120.net ��������
�ǻ���Ƥ�� ������
�ǻ���Ƥ�����ٰ������ࣻƤ֬����
http://16195.dis.999120.net ��������
��״�� ������
��״����verrucacarcinoma
http://16196.dis.999120.net ��������
��Ƥ�ɽ��Լ�Ƥ�� ������
��Ƥ�ɽ��Լ�Ƥ��
http://16197.dis.999120.net ��������
����ϸ���� ������
����ϸ������basalcellepithelioma������ϸ����Ƥ����basaloma��basiloma��BCC��BCE��carcinomabasocellulare��hair-matrixcarcinoma��������������Ƥϸ������������ϸ��������ʴ������Basalcelltumor������ϸ������ͷ�沿����ϸ����
http://16198.dis.999120.net ��������
��״ϸ���� ������
��״ϸ������epidermoidcarcino��pricklecellcarcinoma��squamouscancer��squamouscarcinoma����ƽϸ��������Ƥ����������������ϸ�������۰�����״��Ƥϸ��������ƽϸ����epidermoidcarcino����ϸ����
http://16199.dis.999120.net ��������
��� ������
ë������ë����Ƥ����ë����
http://16200.dis.999120.net ��������
ë��ϸ���� ������
ë��ϸ����
http://16201.dis.999120.net ��������
��� ������
ë������ë��״����ë����
http://16202.dis.999120.net ��������
ë����Χ��ά�� ������
ë����Χ��ά��
http://16203.dis.999120.net ��������
��άë���� ������
��άë������fibrofolliculoma
http://16204.dis.999120.net ��������
������ë�Ҵ����� ������
������ë�Ҵ�������generalizedhairfolliclehamartoma��������ë��������
http://16205.dis.999120.net ��������
ë���ʰ� ������
ë���ʰ�
http://16206.dis.999120.net ��������
ë��Ƥ֬�����Դ����� ������
ë��Ƥ֬�����Դ�������ë��Ƥ֬������������
http://16207.dis.999120.net ��������
Ƥ֬������ ������
Ƥ֬��������adenomasebaceum��Ƥ֬������Pringle��sdisease
http://16208.dis.999120.net ��������
Ƥ֬����Ƥ�� ������
Ƥ֬����Ƥ��
http://16209.dis.999120.net ��������
Ƥ֬�ٰ� ������
Ƥ֬�ٰ���sebaceousadenocarcinoma
http://16210.dis.999120.net ��������
ë��©������ ������
ë��©������
http://16211.dis.999120.net ��������
��ͷ״�������� ������
��ͷ״����������hirdradenomapapilliferum����ͷ״��������syringocystadenomapapilliferum
http://16212.dis.999120.net ��������
����ͷ���������� ������
����ͷ����������
http://16213.dis.999120.net ��������
��ͷ��ʴ�������� ������
��ͷ��ʴ������������ͷ������������
http://16214.dis.999120.net ��������
������ ������
��������eccrinesyringocystadenoma��eruptivahidroadenoma��syringocysadenoma��syringocystoma��syringo-hidroadenoma�������Ժ���������״�����������ܺ���������������������������������������С���ٺ�����
http://16215.dis.999120.net ��������
������������ ������
��������������mixedtumoroftheskin��Ƥ���������������������������Ƥ������������
http://16216.dis.999120.net ��������
���ٰ� ������
���ٰ���apocrineglandcarcinoma��carcinomaofapocrineglands�����������ٰ��������ٰ�
http://16217.dis.999120.net ��������
���������������� ������
������������������Ƥ�����Ի��������Ϯ����������������ת�������Ǻ�����
http://16218.dis.999120.net ��������
���������������� ������
������������������������������
http://16219.dis.999120.net ��������
��ͷ״С�������� ������
��ͷ״С��������
http://16220.dis.999120.net ��������
��ϸ���������� ������
��ϸ������������eccrineacrospiroma��eccrinesweetglandadonomaoftheclearcelltype��nodularhidradenoma��solidcystichidradenoma������Ժ�����������Ժ���������ʵ�������Ժ���������ϸ������������ϸ��С����������С����ĩ�˺�����
http://16221.dis.999120.net ��������
����� ������
�����
http://16222.dis.999120.net ��������
����С���ٺ����� ������
����С���ٺ�������porocarcinoma�����װ�
http://16223.dis.999120.net ��������
С���ٺ�����ά���� ������
С���ٺ�����ά����
http://16224.dis.999120.net ��������
���ܼ�Ƥ�� ������
���ܼ�Ƥ��
http://16225.dis.999120.net ��������
С����Ѫ�ܴ����� ������
С����Ѫ�ܴ�������С����Ѫ�����Դ�����
http://16226.dis.999120.net ��������
�����ٰ� ������
�����ٰ���sclerosingsweatductcarcinoma��Ӳ�����ٵ��ܰ���Ӳ�����ٹܰ�
http://16227.dis.999120.net ��������
��Ϯ��ָֺ��ͷ״�ٰ� ������
��Ϯ��ָֺ��ͷ״�ٰ���aggressivedigitalpolypoidadenocarcinoma����Ϯ��ָֺ��ͷ�ٰ�����Ϯ��ָֺϢ��״�ٰ�
http://16228.dis.999120.net ��������
��״С���ٰ� ������
��״С���ٰ�
http://16229.dis.999120.net ��������
�Һ��С���ٰ� ������
�Һ��С���ٰ���ճ����С������
http://16230.dis.999120.net ��������
Ƥ���������� ������
Ƥ������������Ƥ������������
http://16231.dis.999120.net ��������
��������� ������
�����������������������
http://16232.dis.999120.net ��������
���ٰ� ������
���ٰ���malignantclearcellhidradenoma�����Խ���Ժ�������������ϸ����������������ϸ��ĩ����������
http://16233.dis.999120.net ��������
���Dz��еĶ��ë���� ������
���Dz��еĶ��ë������Cowden���еĶ��ë������Cowden���еĶ��ë�����������Դ������ۺ��������Dz��еĶ��ë�������
http://16234.dis.999120.net ��������
����������� ������
������ë��������pilartumorofthescalp��proliferatingtrichilemmaltumor��ͷ��ë��������ͷƤë������������ëĤ����������ë������������ë�������
http://16235.dis.999120.net ��������
ë����Ƥ�� ������
ë����Ƥ�������������Ƥ����������������Ƥ����pithehomaadenoideysticum��trichofibroacanthoma��trichofibroepithelioma��ë����ά��Ƥ����ë����ά��Ƥ��
http://16236.dis.999120.net ��������
�����֯������ë����Ƥ�� ������
�����֯������ë����Ƥ��
http://16237.dis.999120.net ��������
ëĸ���� ������
ëĸ������calcifiedepithelioma��infundibulo-matrixtumor��trichomatricoma���ƻ���Ƥ�����״�ϸ������ë��������ë��©��-ëĸ������infundibulo-matrixtumor��Malherbescalcifyingepithelioma��Malherbe�ƻ�����Ƥ����pilomatrixoma�����ղ��ƻ���Ƥ�������ղ��ϸƻ���Ƥ��
http://16238.dis.999120.net ��������
Ƥ����ά�� ������
Ƥ����ά����desmoidtumor���ʹ��������ʹ�����ά����Ӳ��ά����fibroushistiocytoma��hardfibroma��nodularsubepidermalfibrosis��sclerosinghemangioma������Ա�Ƥ����ά���ԣ�����Ա�Ƥ����ά������ά����֯ϸ������Ӳ����Ѫ����
http://16239.dis.999120.net ��������
�������� ������
��������
http://16240.dis.999120.net ��������
��Ƥ������ ������
��Ƥ��������sarcomaepithelioides
http://16241.dis.999120.net ��������
Ѫ����ά�� ������
Ѫ����ά����angiofibromas��hemangiofibroma
http://16242.dis.999120.net ��������
�ǵ�����ά��ɫ�� ������
�ǵ�����ά��ɫ�����ǵ�������ά�������ǵ�������ά��ɫ��
http://16243.dis.999120.net ��������
�������ҺѪ����ά�� ������
�������ҺѪ����ά��
http://16244.dis.999120.net ��������
���������� ������
������������Dupuytren������Ledderhosc����palmarandplantarfibromatosisoles��plantarandpalmarfibromatosis
http://16245.dis.999120.net ��������
������������ ������
������������������������������
http://16246.dis.999120.net ��������
�۰� ������
�۰���cancerofcicatrix��Marjolin����������
http://16247.dis.999120.net ��������
���������� ������
������������multipleidiopathichemorrhagicsarcoma������ط��Գ�Ѫ������������������������ϣ�������ط��Զ�Գ�Ѫ���ط��Զ��ɫ�س������������������ɫ�����ط��Գ�Ѫ������
http://16248.dis.999120.net ��������
Ƥ ������
����ά����acrochordon��acrochordons��cutaneoustags��fibomamolle��infantaleperianalpyramidalprotrusions��papillomacolli��skintag��������ͷ����Ƥ�������ࣻ������������ά�������룻Ӥ�����ܽ���¡��Ƥ����ͷ��
http://16249.dis.999120.net ��������
���ʾ�ϸ���� ������
���ʾ�ϸ����
http://16250.dis.999120.net ��������
¡ͻ��Ƥ����ά���� ������
¡ͻ��Ƥ����ά������progressiverecurringdermatofibroma��sarcomatoidfibromaofskin�������Ը�����Ƥ����ά����¡��Ƥ����ά������Ƥ����������ά��
http://16251.dis.999120.net ��������
�Һ��ά���� ������
�Һ��ά������fibrosarcoma��myxosarcoma���Һ������ճҺ��ά����
http://16252.dis.999120.net ��������
������ά��֯ϸ���� ������
������ά��֯ϸ������malignantfibroushistiocytoma��malignanthistiocytoma������ά��֯ϸ������������֯ϸ����
http://16253.dis.999120.net ��������
����Ӳ��֢ ������
������ά�Ժ������ף�indurationofpenis��Peyronie��������������������ף���������ȻӲ�������ά����������Ӳ��֢��penilefibromatosis
http://16254.dis.999120.net ��������
֫����ά�ǻ��� ������
֫����ά�ǻ�����acguireddigitalfibrokeratoma�������ָ(ֺ)��ά�ǻ�����acquiredfibrokeratomaoffinger(toe)��acralibrokeratoma
http://16255.dis.999120.net ��������
Ƥ���Һ�� ������
Ƥ���Һ����Ƥ��ճҺ��
http://16256.dis.999120.net ��������
ëϸѪ���� ������
������Ѫ������������Ѫ����������Ѫ������hemangiomacongenitale��hemangiomasimplex��Kasadach-Merritt�ۺ�����Klippel-Trenaunay�ۺ�����Maffucci�ۺ�������ݮ��Ѫ��������ݮ״Ѫ�������Ƿʴ����ʺ���룻����״Ѫ�������������Σ���ɫ��Ƥ�����룻ëϸѪ�������룻ëϸѪ���������Ѿ�
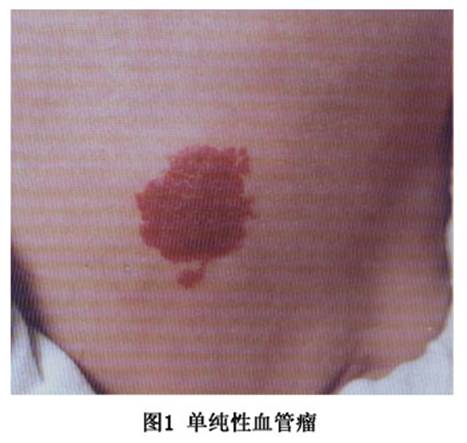
http://16257.dis.999120.net ��������
��������Ѫ���س�����Ѫ���� ������
��������Ѫ���س�����Ѫ����
http://16258.dis.999120.net ��������
����ϸ��Ѫ����Ƥϸ���� ������
����ϸ��Ѫ����Ƥϸ����������ϸ��Ѫ����Ƥ��
http://16259.dis.999120.net ��������
��Ƥ��Ѫ����Ƥϸ���� ������
��Ƥ��Ѫ����Ƥϸ������histiocytoidhemangioma����Ƥ��Ѫ����Ƥ������֯ϸ����Ѫ����
http://16260.dis.999120.net ��������
Ѫ������ͷ״Ѫ����Ƥϸ���� ������
Ѫ������ͷ״Ѫ����Ƥϸ������Dabskatumor��endovascularpapillaryhemangioendothelioma
http://16261.dis.999120.net ��������
Ѫ������ ������
����Ѫ����Ƥϸ������angiosarcoma������Ѫ����Ƥ����Ѫ��������malignanthemangioendothelioma��malignanthomangioendothelioma�������ܰ�Ѫ������
http://16262.dis.999120.net ��������
��״�ܰ��� ������
��״�ܰ����������ܰ�������״ˮ����lymphangiomacysticum��lymphocele���ܰ����ף���������״ˮ��
http://16263.dis.999120.net ��������
����״�ܰ��� ������
����״�ܰ�����cavernomalymphaticum��lymphangiomacavernosum
http://16264.dis.999120.net ��������
ëϸ�ܰ��� ������
ëϸ�ܰ�����locallymphangioma��simplelymphangioma���������ܰ����������ܰ���
http://16265.dis.999120.net ��������
������Ѫ���� ������
������Ѫ����
http://16267.dis.999120.net ��������
��״Ѫ���� ������
��״Ѫ����
http://16268.dis.999120.net ��������
��״Ѫ���� ������
��״Ѫ������angioblastoma��angiomaplexiform��tuftedangioma����Ѫ��ϸ������plexiformangioma����״Ѫ�ܲ�
http://16269.dis.999120.net ��������
Ѫ�ܽǻ��� ������
Ѫ�ܽǻ�����ëϸ���������ࣻѪ�ܽ�������Ѫ����������
http://16270.dis.999120.net ��������
Ѫ������ ������
Ѫ��������Ѫ��ƽ����������Ѫ��������Ѫ��������neuromyoarterialglomustumor
http://16271.dis.999120.net ��������
������Ѫ���� ������
������Ѫ������arteriovenousaneurysm��arteriovenousangioma��������������������Ѫ����
http://16272.dis.999120.net ��������
Ѫ����ϸ���� ������
Ѫ����ϸ������Ѫ����Χϸ����
http://16273.dis.999120.net ��������
�ܰ�������� ������
�ܰ����������Ƥ���ܰ��������
http://16274.dis.999120.net ��������
CD30����Ƥ��Tϸ���ܰ��� ������
CD30����Ƥ��Tϸ���ܰ�����ԭ����CD30����Ƥ��Tϸ���ܰ���
http://16275.dis.999120.net ��������
�̷���Ƥ��CD30���Դ�ϸ���ܰ��� ������
�̷���Ƥ��CD30���Դ�ϸ���ܰ���
http://16276.dis.999120.net ��������
Ƥ��Tϸ���ܰ��� ������
Ƥ��Tϸ���ܰ�����֬Ĥ����Tϸ���ܰ���
http://16277.dis.999120.net ��������
��/����T/NKϸ���ܰ��� ������
��/����T/NKϸ���ܰ���
http://16278.dis.999120.net ��������
ԭ����Ƥ����������ϸ�����ܰ��� ������
ԭ����Ƥ����������ϸ�����ܰ�����ԭ����Ƥ����������ϸ���ܰ���
http://16279.dis.999120.net ��������
ԭ����Ƥ������ϸ���� ������
ԭ����Ƥ������ϸ������lymphoplasmacytoidtymphomaofskin��Ƥ����ϸ�����ܰ�ϸ�����ܰ���
http://16280.dis.999120.net ��������
�̷���Ƥ����������ϸ�����ܰ��� ������
�̷���Ƥ����������ϸ�����ܰ������̷���Ƥ����������ϸ���ܰ���
http://16281.dis.999120.net ��������
Ѫ���ڴ�Bϸ���ܰ��� ������
Ѫ���ڴ�Bϸ���ܰ���
http://16282.dis.999120.net ��������
��ϸ���� ������
��ϸ������plasmacelltumor��plasmocytoma��plasmoma
http://16283.dis.999120.net ��������
��Թ����� ������
��Թ�������Kahler�����������������ز����������ϲ�����ϸ��������
http://16284.dis.999120.net ��������
Ƥ���ܰ���ѿ�ײ� ������
Ƥ���ܰ���ѿ�ײ���lymphogranulomatosiscutis��Ƥ���νܽ�Ƥ�������
http://16285.dis.999120.net ��������
Ƥ����Ѫ�� ������
Ƥ����Ѫ��
http://16286.dis.999120.net ��������
����ϸ����Ѫ�� ������
����ϸ����Ѫ��������ϸ����Ѫ��
http://16287.dis.999120.net ��������
�赥��ϸ����Ѫ�� ������
�赥��ϸ����Ѫ����������ϸ����Ѫ������-����ϸ���Ͱ�Ѫ��
http://16288.dis.999120.net ��������
�ڼ�Ƥ�� ������
�ڼ�Ƥ�������������Ժ�����Ƥ�������ؼ�Ƥ�������Ա�Ƥ��ɫ��ϸ������
http://16289.dis.999120.net ��������
���Ժ�ɫ���� ������
���Ժ����������Ժ�ɫ������amelanoticmalignantmelanoma��invasivemalignantmelanoma��malignantmelanomainsitu��malignantmelanomaofskin������������Ƥ�����Ժ�������Ϯ�Զ��Ժ�ɫ��������ɫ���Ͷ��Ժ�ɫ������ԭλ���Ժ�ɫ����
http://16290.dis.999120.net ��������
ɫ���� ������
ɫ���룻nevoeyticnevus��nevuspigmentous������ϸ���룻��ϸ���룻melanocyticnevus��nevocyticnevus�������ϸ���룻pigmentedmole��pigmentednevus
http://16291.dis.999120.net ��������
̥�� ������
̥�ǣ�Mongolianspot�����ߣ����룻�ɹŰߣ�̥�ߣ�̥�룻��Ƥ�ڱ䲡��dermalmelanosis��mongolianmacula��mongolianspot�����ߣ�birthmark
http://16292.dis.999120.net ��������
������ ������
�����룻������Ǽ�����ɫ��
http://16293.dis.999120.net ��������
���� ������
������ȸ�����룻simplefreckle-likenevus�����ӣ�������
http://16294.dis.999120.net ��������
���� ������
�չ���ȸ�����룻lentigosenilis�����룻������ȸ�����룻˥������ɫ�ࣻsenilefreckle-likenevus
http://16295.dis.999120.net ��������
����ȸ������ ������
����ȸ�����룻Hutchinsonȸ�ߣ�malignantfreckle-likenevus����ǰ����������ϸ����������ǰ�ڱ䲡������ȸ��
http://16296.dis.999120.net ��������
����״ϸ���� ������
����״ϸ���룻����ϸ���룻ballooncellnaevus
http://16297.dis.999120.net ��������
�����Ծ�ɫ���� ������
�����Ծ�ɫ���룻congenitalgiantpigmentednevus����ë�룻��ɫ���룻�����Ծ���ɫ���룻gianthairynevus����ɫ����
http://16298.dis.999120.net ��������
���������� ������
���������룻B-K�ۺ����������������ۺ���
http://16299.dis.999120.net ��������
���� ������
���룻�����룻Suttonnevus�����ٲ�
http://16300.dis.999120.net ��������
���� ������
���룻benignmesenchymalmelanoma��blueneuronevus��chromatophoroma��Jadassohn��Tieche���룻melanofibroma��������ά���������룻���Լ���ʺ��������Լ�Ҷ��ɫ������ɫ��ϸ����
http://16301.dis.999120.net ��������
�������� ������
��������
http://16302.dis.999120.net ��������
��������� ������
�����������benignjuvenilemelanoma��spindleandepithelioidnevus��Spitz�룻�����������������������Ƥ��ϸ���룻�����ͺ����������������ͺ���������������Ƥϸ����
http://16303.dis.999120.net ��������
������ ������
�����룻Beckerɫ����ë�룻Becker�룻���˶���
http://16304.dis.999120.net ��������
Ƥ������ ������
Ƥ��������multiplemucosalneuromas��solitarypalisadedencapsulatedneuroma��traumaticneuromas������Ĥ������������դ��״��Ĥ����������������
http://16305.dis.999120.net ��������
÷�˶�ϸ���� ������
÷�˶�ϸ������Merkelϸ������primaryneuroendocrinecarcinomaoftheskin��trabecularcarcinoma��С��״����ԭ����Ƥ�����ڷ��ڰ���С����
http://16306.dis.999120.net ��������
������ ������
��������chordoblastoma��chordocarcinoma��chordoepithelioma��chordosarcoma��notochordoma��������Ƥ�����ɼ���ϸ����������������������Ƥ��
http://16307.dis.999120.net ��������
�����Һ�� ������
�����Һ����neurothekeoma������ճҺ��
http://16308.dis.999120.net ��������
������ ������
��������schwanncelltumor��schwannoma����Ĥ��ά����������ϸ������ѩ������myoschwannoma��neurilemoma��neurolemmoma��peripheralglioma��schwannoglioma��Schwannoma��sheathtumor������������
http://16309.dis.999120.net ��������
Ƥ����Ĥ�� ������
Ƥ����Ĥ����psammoma��ɳ������ɳ����Ĥ��
http://16310.dis.999120.net ��������
����ϸ���� ������
����ϸ����
http://16311.dis.999120.net ��������
�������� ������
����ά������elehantiasisneuromatosa��molcuseumfibrousm��Recklinghausen����VonRecklinghausen����VonRecklinghausenDisease���������ά�����롤���ֺ�ɭ�������ֻ����������ֻ����ϲ�����ά�����ࣻ��Ƥ����������neuroinomatosis��Recklinghausen'sdisease��von
http://16312.dis.999120.net ��������
�������� ������
��������
http://16313.dis.999120.net ��������
������ ������
��������������ϵͳ����
http://16314.dis.999120.net ��������
���������� ������
С������������opticgliomaofchildren����ͯ��������
http://16315.dis.999120.net ��������
������ֳϸ���� ������
������ֳϸ������������̥ϸ������������̥��֯��
http://16316.dis.999120.net ��������
С��Ѫ����״ϸ���� ������
С��Ѫ����״ϸ������angioreticulomaofchildren��pediatricangioreticuloendothelioma��С��Ѫ����֯ϸ������С��Ѫ����״��Ƥ����С��Ѫ����֯��Ƥ��
http://16318.dis.999120.net ��������
�����Դ����� ������
�����Դ���������������Ԫ������
http://16319.dis.999120.net ��������
���������� ������
������������pediatricspinalcordtumor����������������������
http://16320.dis.999120.net ��������
С������Ĥĸϸ���� ������
С������Ĥĸϸ������pediatricretinalneuroblastoma��retinoblastomaofchildren��С��������Ĥϸ������С������Ĥ����ϸ������pediatricretinalglioblastoma��pediatricretinoma��С������Ĥ���Խ�����
http://16321.dis.999120.net ��������
��ĸϸ���� ������
��ĸϸ����������ϸ����
http://16322.dis.999120.net ��������
С���ȸ�ϸ���� ������
С���ȸ�ϸ������pediatricchromophiletumor��pheochromocytomaofchildren��pediatricchromaffin-celltumor��pediatricchromaffinoma��pediatricmedullarychromaffinoma��pediatricmedullosuprarenoma��pediatricphaeochromocytoma��С����������С����������������С���ȸ�ĸ
http://16323.dis.999120.net ��������
С����ĸϸ���� ������
С����ĸϸ������pediatricembryomaofkidney��С��Willms'����С������̥����С������ϸ������С������������pediatricWhilmstumor��pediatricWilmtumor��pediatricWilmtumorofkidney��pediatricWilmstumor
http://16324.dis.999120.net ��������
С���ʹ��� ������
С���ʹ�����pediatriccraniopharyngealducttumor��pediatrichypophyseal-ducttumor��pediatricRathke'stumor��pediatricsuprasellarcyst��С�����������С�����������ף�С�����ؿ�����С�����ؿ�����
http://16325.dis.999120.net ��������
����֯���� ������
����֯������soft-tissuesarcoma������֯������
http://16327.dis.999120.net ��������
�ݸ��������� ������
�ݸ���������
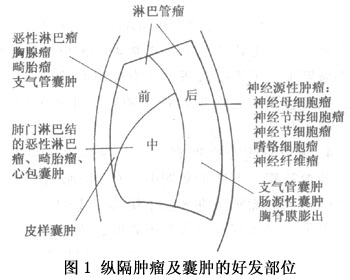
http://16328.dis.999120.net ��������
�������� ������
����������������
http://16329.dis.999120.net ��������
С����ֳϸ���� ������
С����ֳϸ������germcelltumorofchildren��pediatricgerminaltumor��pediatricgerminoma��pediatricgonioma��С����ֳϸ����������pediatricgermcelltumour��С������֯��
http://16330.dis.999120.net ��������
���������� ������
������������pediatrictumorEwing������������������������
http://16331.dis.999120.net ��������
С��ת���Թ����� ������
С��ת���Թ�������С����ת����
http://16332.dis.999120.net ��������
���������� ������
С������������pediatricepostoma��pediatricexos.��pediatricexostosis��pediatrichamartoma��С����������С�����������������ࣻС�����ࣻС���������ࣻpediatricosteoenchondroma
http://16333.dis.999120.net ��������
�����Թ����� ������
�����Թ����ף�unicameralbonecyst�������Թ�����
http://16334.dis.999120.net ��������
���������������� ������
С���������������ף�С������������������
http://16335.dis.999120.net ��������
С����ĸϸ���� ������
С����ĸϸ������medulloblastomaofchildren��С������ϸ����
http://16336.dis.999120.net ��������
�������� ������
С����������pediatricosteogenicsarcoma��pediatricosteomasarcomatosum��С���ɹ�������С����Դ������
http://16337.dis.999120.net ��������
�ѳ����� ������
�ѳ�������tumoroftheovary
http://16338.dis.999120.net ��������
����״���� ������
����״������sarcomabotryoides��sarcomacolliuterihydropicumpapillare
http://16339.dis.999120.net ��������
С����ĸϸ���� ������
С����ĸϸ������С���ζ�ϸ������С������ϸ����
http://16340.dis.999120.net ��������
��β����̥�� ������
��β����̥����sacrococcygealteratoma
http://16341.dis.999120.net ��������
С��Ѫ���� ������
С��Ѫ������hemangiomaofchildren��pediatricangioma��pediatricangioneoplasm��pediatrichaemangioma��pediatrichemartoma��pediatricvasculartumor
http://16342.dis.999120.net ��������
С����ڲ� ������
С����ڲ���Boeck'ssarcoidofchildren��Hutchinson-Boeckdiseaseofchildren��sarcoidosisofchildren��С��������������С����������������С����-�����ϲ���С������������С������״������С������������С����״������pediatricBoeck'ssarcoid��pediatricHutchinson-Boeck
http://16343.dis.999120.net ��������
�����ﲡ ������
�����ﲡ��Fabry-Anderson�����������ϲ����Ѳ�����֬�����ۺ�����������Ѫ�ܽ��������Ŵ���Ӫ��������֬����֢��alpha-galactosidaseAdeficiency��Andeson-Fabrydisease��angiokeratomacorporisdiffusmsyndrome����-��������øAȱ����������ɭ-�����ﲡ����������Ѫ�ܽ�����
http://16344.dis.999120.net ��������
����ڷ����������� ������
����ڷ����������ͣ����Ͷ���ڷ�����������Sipplesyndrome��Sipple�ۺ������������ȸ�ϸ����������״��������-�ȸ�ϸ�����ۺ������������ۺ�����ѩ���ۺ���
http://16345.dis.999120.net ��������
С����״�ٰ� ������
С����״�ٰ���pediatricstrumamaligna����ͯ��״�ٰ���С�����Լ�״���ף�pediatricthyroidcarcinoma
http://16346.dis.999120.net ��������
С������ϸ���� ������
С������ϸ������pediatricastrocyticglioma��С����ϸ������pediatricastroma
http://16347.dis.999120.net ��������
С������ά���� ������
С������ά������pediatricneurocutaneoussyndrome��pediatricRecklinghausen'sdisease��С���������ά������С�����ֻ�������С�����ֻ����ϲ�
http://16348.dis.999120.net ��������
������� ������
С�������С���νܽ�С���νܽ��ϲ���С���ܰ���״ϸ����
http://16349.dis.999120.net ��������
С���ǻ�����ܰ��� ������
С���ǻ�����ܰ�����С�������ܰ�����С���Ǻνܽ��ܰ�����С���Ǻνܽ����ܰ�����С���ǻ�����ܰ�������С���ǻ��������ܰ�����
http://16350.dis.999120.net ��������
С�������ܰ�ϸ����Ѫ�� ������
С�������ܰ�ϸ����Ѫ������ͯ���Գ��ܰ�ϸ����Ѫ������ͯ�����ܰ�ϸ����Ѫ����pediatricacutelymphocyticleukaemia��pediatricacutelymphocyticleukemia��С�����Գ��ܰ�ϸ����Ѫ��
http://16351.dis.999120.net ��������
С�����Է��ܰ�ϸ����Ѫ�� ������
С�����Է��ܰ�ϸ����Ѫ������ͯ�����ܣ���ͯ���Է��ܰ�ϸ����Ѫ����С��������
http://16352.dis.999120.net ��������
С������������Ѫ�� ������
С������������Ѫ����С��������ϸ��δ�ֻ��Ͱ�Ѫ����С��������ϵ��Ѫ����С��������ϸ����Ѫ����С��������ϸ������Ѫ��
http://16353.dis.999120.net ��������
��������-����ϸ����Ѫ�� ������
��������-����ϸ����Ѫ����juvenilechronicmyelogenousleukemia������������ϸ����Ѫ����Ӥ������7�ۺ�����������������ϸ����Ѫ������������-����ϸ���Ͱ�Ѫ��
http://16354.dis.999120.net ��������
С�����Ѫ�� ������
С�����Ѫ����pediatricerythrocytosis��pediatricglobulism��pediatricpolycythemia����ͯ���Ժ�ϸ������֢��С��DiGuglielmo�ۺ�����С����ϸ������֢��С����Ѫ�����ࣻС����Ѫ�Թ�������֢��С�������ܰ�Ѫ����С�����Ժ�ϸ�����財��С����ͯ���Ժ�ϸ������֢
http://16355.dis.999120.net ��������
С��������ϸ����Ѫ�� ������
С��������ϸ����Ѫ����chronicgranulocyticleukemiaofchildhood��chronicmyelogenousleukemiaofchildhood��С��������С��������ϸ����Ѫ����С��������ϸ����Ѫ����pediatricchronicgranulocyticleukemia��pediatricchronicmyelogenousleukemia
http://16356.dis.999120.net ��������
������������ϸ����Ѫ�� ������
������������ϸ����Ѫ����С����������Ѫ����С���Ǽ�������Ѫ����Ӥ����������ϸ����Ѫ��
http://16357.dis.999120.net ��������
������������ϸ����Ѫ�� ������
������������ϸ����Ѫ��
http://16358.dis.999120.net ��������
Ӥ����ά������ ������
Ӥ����ά��������Ӥ����ά�Դ�����
http://16359.dis.999120.net ��������
������Ӥ����ά���� ������
������Ӥ����ά����
http://16360.dis.999120.net ��������
��Ϯ��Ӥ����ά���� ������
��Ϯ��Ӥ����ά������������Ӥ����ά����
http://16361.dis.999120.net ��������
Ӥ��ָֺ��ά���� ������
Ӥ��ָֺ��ά����
http://16362.dis.999120.net ��������
Ӥ������ά���� ������
Ӥ������ά������congenitalgeneralizedfibromatosis�������Է�������ά������������ȫ������ά����
http://16363.dis.999120.net ��������
�������������� ������
��������������
http://16364.dis.999120.net ��������
�����ҹ�Ĥ�� ������
�����ҹ�Ĥ����ependymomaoffossacraniiposterior�������ҹ�Ĥ���������ҹ�Ĥϸ����
http://16365.dis.999120.net ��������
Ӥ������ϸ���� ������
Ӥ������ϸ������Ӥ����ĸϸ����
http://16366.dis.999120.net ��������
������������ ������
С����������������ͯ��������������ͯ��������������С��������������
http://16367.dis.999120.net ��������
�Ըɽ����� ������
�Ըɽ�������neurogliocytomaofbrainstem��neurospongiomaofbrainstem���Ը����������Ը�����ϸ����
http://16368.dis.999120.net ��������
С���ɹ��������� ������
С���ɹ���������
http://16369.dis.999120.net ��������
С���������ͷ״�� ������
С���������ͷ״����pediatricchoroidplexuspapilloma
http://16370.dis.999120.net ��������
��̥�� �����
��̥����dysembryoma��teratoidtumor
http://16371.dis.999120.net ��������
Ƥ����Ƥ������ �����
Ƥ����Ƥ�����ף���Ƥ�����������������Ե�֬������Ƥ������
http://16372.dis.999120.net ��������
��̥�� ������
��̥����dysembryoma��teratoidtumor
http://16373.dis.999120.net ��������
Ƥ����Ƥ������ ������
Ƥ����Ƥ������
http://16374.dis.999120.net ��������
�����Ա��� ���Ǻ�
�����Ա��ף�����ǰ���ף����Ա��ף�rhinitissiccaanterior
http://16376.dis.999120.net ��������
�����Ա��� ���Ǻ�
�����Ա��ף���֬�Ա��ף��ǵ�֬��
http://16377.dis.999120.net ��������
�����Ա��� ���Ǻ�
��̬��Ӧ�Ա��ף���Ӧ�Ա��ף�����֢�������Ա���
http://16378.dis.999120.net ��������
Ѫ���˶��Ա��� ���Ǻ�
Ѫ���˶��Ա��ף�Ѫ�������Ա���
http://16379.dis.999120.net ��������
���и�ƫ�� ���Ǻ�
���и�ƫ�������и�����
http://16380.dis.999120.net ��������
���и�Ѫ�� ���Ǻ�
���и�Ѫ��
http://16381.dis.999120.net ��������
���и�ŧ�� ���Ǻ�
���и�ŧ�ף�nasoseptalabscess
http://16382.dis.999120.net ��������
���и����� ���Ǻ�
�������
http://16383.dis.999120.net ��������
��ǻ��������� ���Ǻ�
��ǻ���������
http://16384.dis.999120.net ��������
��ͯ����� ���Ǻ�
��ͯ�����
http://16385.dis.999120.net ��������
�����ǿ�Ϣ�� ���Ǻ�
�����ǿ�Ϣ��
http://16386.dis.999120.net ��������
���� ���Ǻ�
���ܣ����ۣ��Ǵ�
http://16387.dis.999120.net ��������
������� ���Ǻ�
�������
http://16388.dis.999120.net ��������
��ǰͥ���� ���Ǻ�
��ǰͥ����
http://16389.dis.999120.net ��������
����Һ���� ���Ǻ�
����Һ����
http://16390.dis.999120.net ��������
��Һ���� ���Ǻ�
��Һ����
http://16391.dis.999120.net ��������
������Դ������ ���Ǻ�
������Դ������
http://16392.dis.999120.net ��������
����ͷ״�� ���Ǻ�
����ͷ״����chneiderain��ͷ״������״ϸ����ͷ״������������ͷ״��
http://16393.dis.999120.net ��������
��ǻ������������ ���Ǻ�
��ǻ������������
http://16394.dis.999120.net ��������
�Ƕ�����ѿ���ܰ��� ���Ǻ�
�Ƕ�����ѿ���ܰ�������ǻ����ܰ���
http://16395.dis.999120.net ��������
�����ѹ�� ���Ǻ�
�����ѹ�ˣ���ѹ�����Ա���ף����ձ����
http://16396.dis.999120.net ��������
��ǰͥʪ�� ���Ǻ�
��ǰͥʪ��
http://16397.dis.999120.net ��������
������ ���Ǻ�
�����ǣ��ư���������������
http://16398.dis.999120.net ��������
���Ա��� ���Ǻ�
���Ա��ף��˷磻��ͨ��ð�������Ա���
http://16399.dis.999120.net ��������
���Ե����Ա��� ���Ǻ�
���Ե����Ա��ף����Ա���
http://16400.dis.999120.net ��������
���Էʺ��Ա��� ���Ǻ�
���Էʺ��Ա���
http://16401.dis.999120.net ��������
ҩ���Ա��� ���Ǻ�
ҩ���Ա��ף��ж��Ա���
http://16402.dis.999120.net ��������
ή���Ա��� ���Ǻ�
ή���Ա��ף�����֢��ozena
http://16403.dis.999120.net ��������
���Ա������� ���Ǻ�
���Ա������ף�������������ף���Ͽ�ף������ͣ����
http://16404.dis.999120.net ��������
÷���� ���Ǻ�
�����֢��÷���������֢���ʺ��������У������ۺ������ʺ���֢������ۺ�����globushystericus
http://16405.dis.999120.net ��������
�ʲ����� ���Ǻ�
�ʲ�����
http://16406.dis.999120.net ��������
�ʲ����� ���Ǻ�
�ʲ�����
http://16407.dis.999120.net ��������
������˯�ߺ�����ͣ�ۺ��� ���Ǻ�
������˯�ߺ�����ͣ�ۺ�����ƥ������(Pickwickian)�ۺ���
http://16409.dis.999120.net ��������
������άѪ���� ���Ǻ�
������άѪ�����������ഺ�ڳ�Ѫ�Ա���Ѫ����ά��
http://16410.dis.999120.net ��������
�ʲ������� ���Ǻ�
�ʲ�������
http://16411.dis.999120.net ��������
�ʹ��� ���Ǻ�
�ʹ��������ؿ���
http://16412.dis.999120.net ��������
���ʰ� ���Ǻ�
���ʰ�
http://16413.dis.999120.net ��������
������������� ���Ǻ�
�������������
http://16414.dis.999120.net ��������
���Ա������� ���Ǻ�
���Ա�������
http://16415.dis.999120.net ��������
���ʶ������� ���Ǻ�
���ʶ������������ʶ�������
http://16416.dis.999120.net ��������
����ؽ������ۺ��� ���Ǻ�
����ؽ������ۺ���
http://16417.dis.999120.net ��������
������ʴ� ���Ǻ�
������ʴ�
http://16418.dis.999120.net ��������
��������ŧ�� ���Ǻ�
��������ŧ�ף���Ӹ
http://16419.dis.999120.net ��������
�ʺ�ŧ�� ���Ǻ�
�ʺ�ŧ��
http://16420.dis.999120.net ��������
����ŧ�� ���Ǻ�
����ŧ��
http://16421.dis.999120.net ��������
�ʸо����˻�ȱʧ ���Ǻ�
�ʸо����˻�ȱʧ
http://16422.dis.999120.net ��������
������ʹ ���Ǻ�
������ʹ
http://16423.dis.999120.net ��������
�����Ժ��� ���Ǻ�
�����Ժ���
http://16425.dis.999120.net ��������
�����Ժ�����֢ ���Ǻ�
�����Ժ�����֢
http://16426.dis.999120.net ��������
��Դ���������� ���Ǻ�
��Դ���������ܣ��������ף��ܰ���Ƥ���ף���������
http://16427.dis.999120.net ��������
����ӵ�� ��ǻ��
����ӵ��������ӵ����crowdingofteeth
http://16428.dis.999120.net ��������
ȣ�� ��ǻ��
ȣ��ȣ�ݣ�ȣ����ȣ��
http://16429.dis.999120.net ��������
��ǻ�������� ��ǻ��
��ǻ��������
http://16430.dis.999120.net ��������
���Ա�Ե������ ��ǻ��
���Ա�Ե�����ף����������ף�������Ե�ף���Ե�ף�simplegingivitis��marginalgingivitis
http://16431.dis.999120.net ��������
�dzݹ����� ��ǻ��
������ĥ�������ף��dzݹ����ף�pericoronitisofwisdomtooth
http://16432.dis.999120.net ��������
�沿��Ӹ ��ǻ��
�沿��Ӹ
http://16433.dis.999120.net ��������
�澱����ŧ���ܰͽ��� ��ǻ��
�澱����ŧ���ܰͽ��ף�facialandcervicalsuppurativelymphadenitis
http://16434.dis.999120.net ��������
���¼�϶��Ⱦ ��ǻ��
���¼�϶��Ⱦ�����¼�϶����֯�ף����¼�϶������֯�ף�infraorbitalspaceinfection
http://16435.dis.999120.net ��������
��¼�϶��Ⱦ ��ǻ��
��¼�϶��Ⱦ����¼�϶������֯�ף���¼�϶����֯�ף�submentalspaceinfection
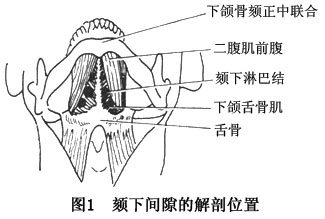
http://16436.dis.999120.net ��������
�ڵ��϶��Ⱦ ��ǻ��
�ڵ��϶��Ⱦ���ڵ�����֯�ף��ڵ���֯�ף�mouthfloorcellulitis��Cellulitisoffloorofmouth
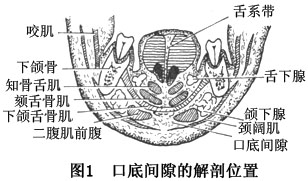
http://16437.dis.999120.net ��������
�ռ�϶��Ⱦ ��ǻ��
�ռ�϶��Ⱦ���ռ�϶������֯�ף��ռ�϶����֯�ף�buccalspaceinfection
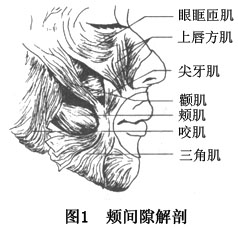
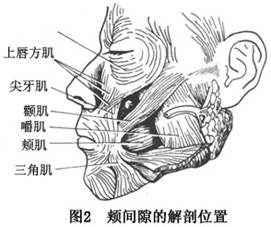
http://16438.dis.999120.net ��������
��϶��Ⱦ ��ǻ��
��϶��Ⱦ����϶����֯�ף���϶������֯�ף�temporalspaceinfection
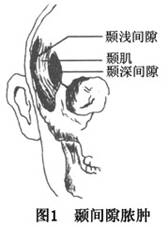
http://16439.dis.999120.net ��������
��¼�϶��Ⱦ ��ǻ��
��¼�϶��Ⱦ����¼�϶������֯�ף���¼�϶����֯�ף�infratemporalspaceinfection

http://16440.dis.999120.net ��������
ҧ���¼�϶��Ⱦ ��ǻ��
ҧ���¼�϶��Ⱦ��ҧ����϶��Ⱦ��ҧ���¼�϶������֯�ף�ҧ���¼�϶����֯��
http://16441.dis.999120.net ��������
������϶��Ⱦ ��ǻ��
������϶��Ⱦ��������϶������֯�ף�������϶����֯�ף�pterygomandibularspaceinfection
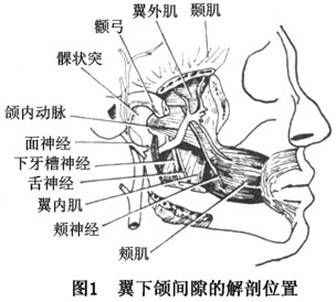
http://16442.dis.999120.net ��������
���¼�϶��Ⱦ ��ǻ��
���¼�϶��Ⱦ�����¼�϶������֯�ף����¼�϶����֯�ף�sublingualspaceinfection
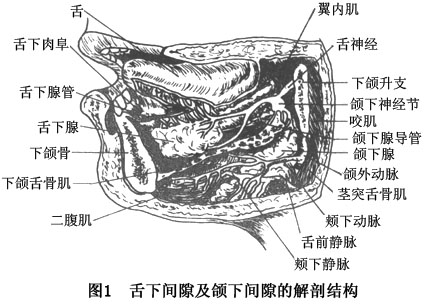
http://16443.dis.999120.net ��������
���Լ�϶��Ⱦ ��ǻ��
���Լ�϶��Ⱦ�����Լ�϶������֯�ף����Լ�϶����֯�ף�lateralpharyngealspaceinfection

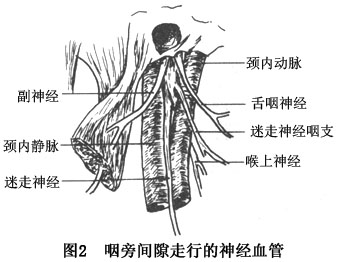
http://16444.dis.999120.net ��������
����¼�϶��Ⱦ ��ǻ��
����¼�϶��Ⱦ����¼�϶������֯�ף���¼�϶����֯�ף�submandibularspaceinfection
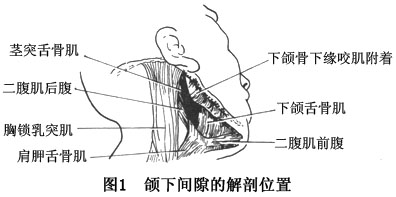
http://16445.dis.999120.net ��������
��ŧ���ǹ����� ��ǻ��
��ŧ���ǹ�����
http://16446.dis.999120.net ��������
Ӥ�����ǹ����� ��ǻ��
Ӥ�����ǹ����ף��������ǹ����ף�ѪԴ���ǹ�����
http://16447.dis.999120.net ��������
�������ǹ����� ��ǻ��
�������ǹ����ף��������ǻ�����osteoradionecrosis
http://16448.dis.999120.net ��������
����Ӳ�����ǹ����� ��ǻ��
����Ӳ�����ǹ����ף����Էǻ�ŧ��Ӳ���Թ����ף�GarreӲ���Թ����ף�Garr�������ף�Garre��ssclerosingosteomyelitis
http://16449.dis.999120.net ��������
��״���� ��ǻ��
��ǻ��״�����״���herpeszoster
http://16450.dis.999120.net ��������
�ഺ������ ��ǻ��
�ഺ�����ף����������ף�pubertalgingivitis
http://16451.dis.999120.net ��������
������ܰͽ��� ��ǻ��
������ܰͽ��ף�lymphadenitistuberculosa
http://16452.dis.999120.net ��������
���ǽ�� ��ǻ��
���ǽ�ˣ�maxillofacialbonetuberculosis
http://16453.dis.999120.net ��������
��沿���߾��� ��ǻ��
��沿���߾�����maxillofacialactinomycosis
http://16454.dis.999120.net ��������
��沿÷�� ��ǻ��
��沿÷����maxillofacialsyphilis
http://16455.dis.999120.net ��������
���������� ��ǻ��
���������ף�����������
http://16456.dis.999120.net ��������
����ڲ� ��ǻ��
��-��-�ڲ�
http://16457.dis.999120.net ��������
��ǻ������� ��ǻ��
��ǻ���������oralcandidiasis
http://16458.dis.999120.net ��������
���Ի������������� ��ǻ��
���Ի������������ף����Ի��������������ף���������������ף�ս���ڣ�Vincent���ף���ɭ���ף�Vincentgingivitis��trenchmouth
http://16459.dis.999120.net ��������
��ǻ��� ��ǻ��
��ǻ��ˣ�oraltuberculosis
http://16460.dis.999120.net ��������
ҩ������������ ��ǻ��
ҩ������������
http://16461.dis.999120.net ��������
������ά���� ��ǻ��
������ά�������Ŵ���������ά������������������ά�������ط���������ά������hereditarygingivalfibromatosis��familialgingivalfibromatosis��idiopathicgingivalfibromatosis
http://16462.dis.999120.net ��������
����Կ��� ��ǻ��
����Կ���
http://16463.dis.999120.net ��������
�����Կ��� ��ǻ��
�����Կ��ף���������ڼջ��ң�noma
http://16464.dis.999120.net ��������
��������ͷ�� ��ǻ��
��������ͷ�ף�����ͷ��
http://16465.dis.999120.net ��������
���Զ����ŧ�� ��ǻ��
���Զ����ŧ��
http://16466.dis.999120.net ��������
����� ��ǻ��
����գ���֢
http://16467.dis.999120.net ��������
��������� ��ǻ��
�������������������֢
http://16468.dis.999120.net ��������
��³���ۺ��� ��ǻ��
��³���ۺ�����Crouzon�ۺ���������ͷ�ۺ����������Լ�ͷ��ָ(ֺ)�����ۺ��������ۺ�����Virchow�ۺ���
http://16470.dis.999120.net ��������
Apert�ۺ��� ��ǻ��
Apert�ۺ�������ͷ��ָ���Σ���ָ�ͼ�ͷ�ۺ�������ͷ��ָ�ۺ�����acrocephalosyndactyly
http://16471.dis.999120.net ��������
���ͻ�ʴ� ��ǻ��
���ͻ�ʴ�
http://16472.dis.999120.net ��������
�������� ��ǻ��
���������������ۺ��������ż�������
http://16473.dis.999120.net ��������
ǰ����he ��ǻ��
ǰ����he
http://16474.dis.999120.net ��������
���������� ��ǻ��
���������ף����Գ��������ף�chronicadultperiodontitis
http://16475.dis.999120.net ��������
ҩ������Կ��� ��ǻ��
ҩ������Կ���
http://16476.dis.999120.net ��������
��ǻ��沿����֯���� ��ǻ��
��ǻ��沿����֯���ˣ�abrasiooftheoromaxillo-facialregionsofttissue
http://16477.dis.999120.net ��������
���ʷ�����ȫ ��ǻ��
���ʷ�����ȫ�����ʷ�����ȫ֢
http://16478.dis.999120.net ��������
������ ��ǻ��
����֢������������������mottledenamel
http://16479.dis.999120.net ��������
�Ļ����� ��ǻ��
�Ļ�����
http://16480.dis.999120.net ��������
�Ŵ��������ʷ�����ȫ ��ǻ��
�Ŵ��������ʷ�����ȫ���Ŵ������������
http://16481.dis.999120.net ��������
������÷���� ��ǻ��
������÷����
http://16482.dis.999120.net ��������
��ǻ��沿����֯���� ��ǻ��
��ǻ��沿����֯���ˣ�contusedwoundoftheoromaxillo-facialregionsofttissue
http://16483.dis.999120.net ��������
��С���������������� ��ǻ��
������������������
http://16484.dis.999120.net ��������
�ں�����˫����������� ��ǻ��
�ں�����˫�����������
http://16485.dis.999120.net ��������
��������� ��ǻ��
���������
http://16486.dis.999120.net ��������
������ ��ǻ��
������
http://16487.dis.999120.net ��������
���� ��ǻ��
����
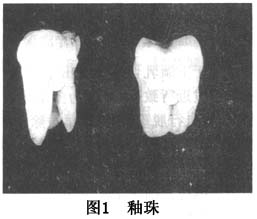
http://16488.dis.999120.net ��������
��ǻ��沿����֯������ ��ǻ��
��ǻ��沿����֯�����ˣ�contusionandlaceratingwoundoftheoromaxillo-facialregionsofttissue
http://16489.dis.999120.net ��������
������ ��ǻ��
��������������
http://16490.dis.999120.net ��������
������ȱ���� ��ǻ��
������ȱ����
http://16491.dis.999120.net ��������
��ǻ��沿����֯�и��� ��ǻ��
��ǻ��沿����֯�и��ˣ�concisusoftheoromaxillo-facialregionsofttissue
http://16492.dis.999120.net ��������
���� ��ǻ��
����
http://16494.dis.999120.net ��������
��ǻ��沿����֯���� ��ǻ��
��ǻ��沿����֯���ˣ�punctureoftheoromaxillo-facialregionsofttissue
http://16495.dis.999120.net ��������
��ǻ��沿����֯˺���� ��ǻ��
��ǻ��沿����֯˺���ˣ�avulsedwoundoftheoromaxillo-facialregionsofttissue
http://16496.dis.999120.net ��������
��ǻ��沿����֯����ҧ�� ��ǻ��
��ǻ��沿����֯����ҧ�ˣ�animalbiteoftheoromaxillo-facialregionsofttissue
http://16497.dis.999120.net ��������
�����ԽӴ��Կ��� ��ǻ��
�����ԽӴ��Կ���
http://16498.dis.999120.net ��������
�������������� ��ǻ��
�������������ף����ܱ��ԣ�periodontosis
http://16499.dis.999120.net ��������
����λ ��ǻ��
�����dislocationoftheteeth
http://16500.dis.999120.net ��������
����ĥ�� ��ǻ��
����ĥ��ĥ��֢��ĥ��
http://16501.dis.999120.net ��������
ĥ��֢ ��ǻ��
ĥ��֢��ҹĥ��֢
http://16503.dis.999120.net ��������
Ш״ȱ�� ��ǻ��
Ш״ȱ��
http://16504.dis.999120.net ��������
���� ��ǻ��
����toothconcussion
http://16505.dis.999120.net ��������
���� ��ǻ��
���ۣ����۶ϣ�Odontoclasia
http://16506.dis.999120.net ��������
��ʴ֢ ��ǻ��
��ʴ֢
http://16507.dis.999120.net ��������
������ ��ǻ��
�����ѣ���ȫ���ѣ����ѣ�imcompletetoothfracture
http://16508.dis.999120.net ��������
�������� ��ǻ��
��������
http://16509.dis.999120.net ��������
���ٽ����������� ��ǻ��
���ٽ����������ף����ٽ�չ��������
http://16510.dis.999120.net ��������
���ǹ��� ��ǻ��
���ǹ��ۣ�maxillaryfracture
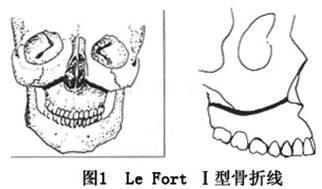
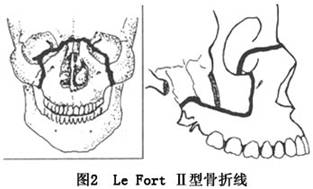
http://16511.dis.999120.net ��������
���ǹ��� ��ǻ��
���ǹ��ۣ�mandibularfracture
http://16512.dis.999120.net ��������
�����ʹ��� ��ǻ��
�����ʹ����������ʹ���֢�����ݸо�����֢�������������ʣ�����������֢
http://16513.dis.999120.net ��������
�ഺǰ�������� ��ǻ��
�ഺǰ��������
http://16514.dis.999120.net ��������
ȧ�Ǽ�ȧ������ ��ǻ��
ȧ�Ǽ�ȧ�����ۣ�zygomaticandarchfracture
http://16515.dis.999120.net ��������
�ǹǹ��� ��ǻ��
�ǹǹ���
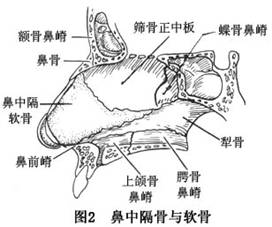
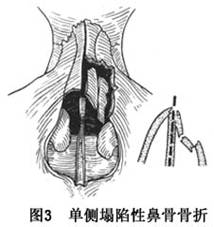
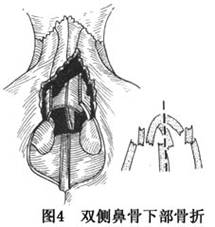
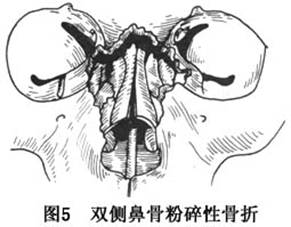
http://16516.dis.999120.net ��������
���Ƚǻ����ܲ��ۺ��� ��ǻ��
���Ƚǻ����ܲ��ۺ�����Papillon-Lefevre�ۺ��������Ƚǻ�-�����ƻ��ۺ�����Papillon-Lefevresyndrome��syndromeofpalmar-plantarhyper-keratosisandprematureperiodontaldestruction
http://16517.dis.999120.net ��������
�����ۺ��� ��ǻ��
�����ۺ�����Down�ۺ������������ͣ�Ⱦɫ��21-�����ۺ�����mongolism
http://16518.dis.999120.net ��������
������������ ��ǻ��
�����������ף�������������
http://16519.dis.999120.net ��������
���������ȱ���ۺ�������������� ��ǻ��
���������ȱ���ۺ�������������ף�HIV����������ף����̲������������
http://16520.dis.999120.net ��������
�ۿ����� ��ǻ��
�ۿ����ۣ�blow-outfracture
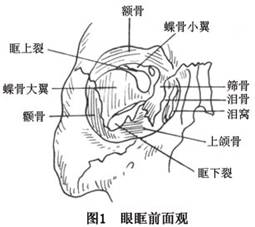
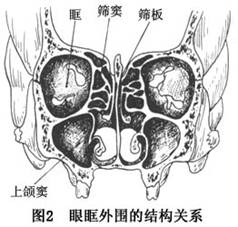

http://16521.dis.999120.net ��������
����������� ��ǻ��
����������ף�������������
http://16522.dis.999120.net ��������
ȫ�沿���� ��ǻ��
ȫ�沿����
http://16523.dis.999120.net ��������
��ǻ��沿������ ��ǻ��
��ǻ��沿�����ˣ�firearminjuryoforomaxillo-facialregion
http://16524.dis.999120.net ��������
��ǻ��沿���� ��ǻ��
��ǻ��沿���ˣ�burnoforomaxillo-facialregion
http://16525.dis.999120.net ��������
��ǻ��沿�������� ��ǻ��
��ǻ��沿�������ˣ�nuclearweaponwoundoforomaxillo-facialregion
http://16526.dis.999120.net ��������
��ǻ��沿��ѧ������ ��ǻ��
��ǻ��沿��ѧ�����ˣ�woundofchemicalweaponoforomaxillo-facialregion
http://16527.dis.999120.net ��������
ǰ����� ��ǻ��
ǰ�����
http://16528.dis.999120.net ��������
�����Կ�ǻ���� ��ǻ��
�����Կ�ǻ�������������Կ�ǻ�������Կڴ����������������ף�recurrentaphthousstomatitis��recurrentaphthousulcer��recurrentoralulcer
http://16529.dis.999120.net ��������
�����������ϲ��� ��ǻ��
�����������ϲ���
http://16530.dis.999120.net ��������
�ɸ��������� ��ǻ��
�ɸ���������
http://16531.dis.999120.net ��������
��״������� ��ǻ��
��״������ף�cystisthyrolingualis
http://16532.dis.999120.net ��������
�������� ��ǻ��
��������
http://16533.dis.999120.net ��������
�������� ��ǻ��
��������
http://16534.dis.999120.net ��������
���������� ��ǻ��
����������
http://16535.dis.999120.net ��������
���������� ��ǻ��
���������ף��������ף�radicularcyst
http://16536.dis.999120.net ��������
ʼ������ ��ǻ��
ʼ������
http://16537.dis.999120.net ��������
�������� ��ǻ��
�������ף��������ף�follicularcyst
http://16538.dis.999120.net ��������
���������� ��ǻ��
����������
http://16540.dis.999120.net ��������
��״������� ��ǻ��
��״�������
http://16541.dis.999120.net ��������
��������� ��ǻ��
��������ף���������ף����������ף����������ף�����ǰ�������
http://16542.dis.999120.net ��������
�������� ��ǻ��
��������
http://16543.dis.999120.net ��������
�Ǵ����� ��ǻ��
�Ǵ�����
http://16544.dis.999120.net ��������
Ѫ���������� ��ǻ��
Ѫ���������ף����������ף������Թ����ף�solitarycyst��traumaticbonecyst
http://16545.dis.999120.net ��������
�������Թ����� ��ǻ��
�������Թ�����
http://16546.dis.999120.net ��������
���軵�� ��ǻ��
���軵���������Ի���
http://16547.dis.999120.net ��������
����ƻ� ��ǻ��
����ƻ�����ʯ
http://16548.dis.999120.net ��������
�������� ��ǻ��
�������գ�internalresorption
http://16549.dis.999120.net ��������
���ֲ没�� ��ǻ��
���ֲ没��
http://16551.dis.999120.net ��������
������ ��ǻ��
��������paradentoma
http://16552.dis.999120.net ��������
���Խ�Һ�Ը������� ��ǻ��
���Խ�Һ�Ը�������
http://16553.dis.999120.net ��������
��������ŧ�� ��ǻ��
���Ի�ŧ�Ը������ף���������ŧ�ף����Ը�����ŧ�ף��������ļ��Ի�ŧ�ڣ�Acutealveolarabscess
http://16554.dis.999120.net ��������
��ϸ����ѿ�� ��ǻ��
��ϸ����ѿ�ף���ϸ��������ѿ�ף�giantcellreparationgranuloma��GCRG
http://16555.dis.999120.net ��������
���Ը������� ��ǻ��
���Ը�������
http://16556.dis.999120.net ��������
���������� ��ǻ��
����������
http://16557.dis.999120.net ��������
�����Կ��� ��ǻ��
�����Կ��ף��������Ĥ�ף�radiatemucositis
http://16558.dis.999120.net ��������
����ŧ�� ��ǻ��
����ŧ��
http://16559.dis.999120.net ��������
����ϸ���� ��ǻ��
����ϸ����������ϸ����
http://16560.dis.999120.net ��������
����ϸ����ά�� ��ǻ��
����ϸ����ά��������ϸ����ά����������������ά�ͳ���ϸ���������Ի��������
http://16561.dis.999120.net ��������
��״��Դ���� ��ǻ��
��״��Դ��������Դ����״ϸ����
http://16563.dis.999120.net ��������
�ƻ���Ƥ����Դ���� ��ǻ��
�ƻ���Ƥ����Դ��������Դ�Ըƻ���Ƥ����pindborg��
http://16565.dis.999120.net ��������
�ƻ�����Դ������ ��ǻ��
�ƻ�����Դ�����ף���Դ�Ըƻ�����
http://16566.dis.999120.net ��������
��Դ����ά�� ��ǻ��
��Դ����ά����calcifyingodontogeniccyst
http://16567.dis.999120.net ��������
��Դ���Һ�� ��ǻ��
��Դ���Һ������Դ��ճҺ��
http://16568.dis.999120.net ��������
�������� ��ǻ��
��������
http://16569.dis.999120.net ��������
���Գ�������ϸ���� ��ǻ��
���Գ�������ϸ����������������ĸϸ������������������
http://16570.dis.999120.net ��������
���� ��ǻ��
����
http://16571.dis.999120.net ��������
��¡ ��ǻ��
��¡���������ࣻ���ࣻexostosis��torus
http://16572.dis.999120.net ��������
�����ʻ���ά�� ��ǻ��
�����ʻ���ά���������ʻ�����ά��
http://16573.dis.999120.net ��������
�����������ʽṹ���� ��ǻ��
�����������ʽṹ��������������ά�ṹ������periapicalfibrousdysplasia
http://16574.dis.999120.net ��������
���������� ��ǻ��
�������������������������������Զ������������familialmultiplecementoma
http://16575.dis.999120.net ��������
���֢ ��ǻ��
���֢���������Ƿʴ���ʹ��������������ά�쳣��ֳ֢��familialcherubism��familialfibrousdysplasiaofthejaws
http://16576.dis.999120.net ��������
��ǻ�Ĥ���������� ��ǻ��
��ǻ�Ĥ������������������ϸ����ѿ�ף�traumaticeosinophilicgranuloma��TEG
http://16577.dis.999120.net ��������
����ή�� ��ǻ��
��������������ή������Ե��֯������marginaltissuerecession��periodontalatrophy
http://16578.dis.999120.net ��������
���� ��ǻ��
������carcinomaofthelip
http://16579.dis.999120.net ��������
��ǻ��沿�����ܰ��� ��ǻ��
��ǻ��沿�����ܰ���
http://16580.dis.999120.net ��������
��ǻ��沿��ϸ������ ��ǻ��
��ǻ��沿��ϸ����������ǻ��沿������
http://16581.dis.999120.net ��������
��ǻ��沿������������ѿ�� ��ǻ��
��ǻ��沿������������ѿ�ף�������ѿ�ף���������ѿ�ף�������������ѿ�ף�midlinelethalgranuloma��necroticgranuloma��malig-nantgranuloma
http://16582.dis.999120.net ��������
��ǻ��沿�ɸ�˹ϸ���� ��ǻ��
��ǻ��沿�ɸ�˹ϸ�������ʸ�˹ϸ����ѿ�ף���֯ϸ������֢X���ط�����֯ϸ������֢��idiopathichistiocytosis��Langerhanscellgranuloma��histiocytosisX
http://16583.dis.999120.net ��������
��ǻ��沿ת������ ��ǻ��
��ǻ��沿ת��������metastaticcarcinomainthemaxillofacialregion
http://16584.dis.999120.net ��������
�ڵװ� ��ǻ��
�ڵװ�
http://16586.dis.999120.net ��������
������ ��ǻ��
������
http://16587.dis.999120.net ��������
�հ� ��ǻ��
�հ���carcinomaofcheek
http://16588.dis.999120.net ��������
Ӳ�� ��ǻ��
Ӳ��
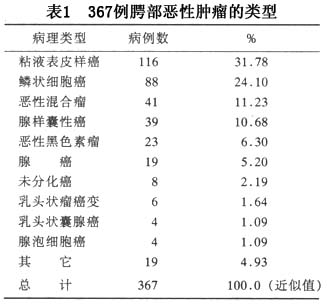
http://16589.dis.999120.net ��������
���ʰ� ��ǻ��
���ʰ���carcinomaoforal-pharynx
http://16590.dis.999120.net ��������
��� ��ǻ��
���
http://16591.dis.999120.net ��������
�������ǰ� ��ǻ��
�������ǰ����������ǰ���ԭ�������ڰ���ԭ���������ڰ���primaryintraosseouscarcinoma��primaryintra-alveolarcarcinoma
http://16592.dis.999120.net ��������
���沿Ƥ���� ��ǻ��
���沿Ƥ����
http://16593.dis.999120.net ��������
��ǻ��沿����֯���� ��ǻ��
��ǻ��沿����֯����
http://16594.dis.999120.net ��������
��ǻ��沿��Դ������ ��ǻ��
��ǻ��沿��Դ������
http://16595.dis.999120.net ��������
��ɫ���� ��ǻ��
��ǻ��沿���Ժ���������ɫ��������ɫ�ذ�
http://16596.dis.999120.net ��������
���״�� ��ǻ��
���״�٣�����λ��״�٣�ectopicthyroid
http://16597.dis.999120.net ��������
������ ��ǻ��
������
http://16599.dis.999120.net ��������
��ֲ����Χ���� ��ǻ��
��ֲ����Χ���䣻��ֲ������֯��֢
http://16600.dis.999120.net ��������
��ǻ��ƽ̦ ��ǻ��
��ǻ��ƽ̦
http://16601.dis.999120.net ��������
�ڸ���֢ ��ǻ��
�ڸ���֢
http://16602.dis.999120.net ��������
���� ��ǻ��
����
http://16603.dis.999120.net ��������
��ǻ��ɫ�ǻ��� ��ǻ��
��ǻ��ɫ�ǻ��������Խǻ���������ɫ�ǻ�����ǰ�װߣ���Ƥ�����̼��Կ��ף��̼���ɫ�ǻ�����benignhyperkeratosis��leukokeratosisnicotinapalati��nicotinicstomatitis
http://16604.dis.999120.net ��������
���Ի�ŧ�������� ��ǻ��
���Ի�ŧ��������
http://16605.dis.999120.net ��������
���������������� ��ǻ��
����������������
http://16607.dis.999120.net ��������
��ʯ������������� ��ǻ��
��ʯ������������ף�����Ӳ����������ף�Kuttner����sialolithiasisandsialadenitisofthesubmandibulargland
http://16608.dis.999120.net ��������
���ٽ�� ��ǻ��
���ٽ�ˣ���Һ�ٽ��
http://16609.dis.999120.net ��������
���ٷ��߾��� ��ǻ��
���ٷ��߾�������Һ�ٷ��߾�����actinomycosisofsalivarygland
http://16610.dis.999120.net ��������
��ǻ�װ� ��ǻ��
��ǻ�װߣ���ǻ�װ�֢
http://16611.dis.999120.net ��������
�������˺����� ��ǻ��
�������˺�����
http://16612.dis.999120.net ��������
��ǻ��� ��ǻ��
��ǻ��ߣ���ǻ��߲�����ֳ�Ժ�ߣ������غ�ߣ���ɫ��ֳ�Բ��䣻��ɫ�ʺ�֢��erythroplasticlesion��erythroplasiaofQueyrat
http://16613.dis.999120.net ��������
�����Һ���� ��ǻ��
�����Һ���ף���Һ���Һ���ף�salivarymucocele
http://16614.dis.999120.net ��������
�������� ��ǻ��
��������
http://16615.dis.999120.net ��������
�������Էʴ� ��ǻ��
�������Էʴ������״�֢����Һ�����Էʴ������������״�sialadenosis
http://16616.dis.999120.net ��������
��ǻ��״����Ǵ� ��ǻ��
��ǻ��״����Ǵ���discoidlupuserythematosus
http://16617.dis.999120.net ��������
���������� ��ǻ��
�������������������mixedtumor��pleomorphicadenoma
http://16618.dis.999120.net ��������
��Ƥ-����Ƥ�� ��ǻ��
��Ƥ-����Ƥ������״ʵ���������ټ���Ƥ��������Ƥϸ���������������������ܰ�
http://16619.dis.999120.net ��������
�����ԵͶȶ����ٰ� ��ǻ��
�����ԵͶȶ����ٰ�����ǻС����Ҷ״�ٰ�����ĩ�����ٰ���terminalductadenocarcinoma��lobularcarcinoma��terminalductcarcinoma
http://16620.dis.999120.net ��������
���Զ��������� ��ǻ��
���Զ���������
http://16621.dis.999120.net ��������
�����ٰ� ��ǻ��
�����ٰ�����Һ���ٰ����������������ٰ�������������Һ�ٰ���adenocarcinomanototherwisespecified
http://16622.dis.999120.net ��������
������״ϸ���� ��ǻ��
������״ϸ��������Һ����״ϸ������
http://16623.dis.999120.net ��������
���ٰ��ܰ������ʵ�δ�ֻ��� ��ǻ��
���ٰ��ܰ������ʵ�δ�ֻ�������Һ�ٰ��ܰ������ʵ�δ�ֻ���
http://16624.dis.999120.net ��������
����Сϸ���� ��ǻ��
����Сϸ��������Һ��Сϸ����
http://16625.dis.999120.net ��������
�ܰ�������ͷ״������ ��ǻ��
�ܰ�������ͷ״�����������ܰ���������������ͷ״�ܰ�������������ͷ״�ܰ���������adenolymphoma��Warthintumor
http://16626.dis.999120.net ��������
���������� ��ǻ��
������������������ϸ������������ϸ����������������ϸ������Oxyphilicgranularcelladenoma��Oncocytoma
http://16627.dis.999120.net ��������
��״���� ��ǻ��
��״����
http://16628.dis.999120.net ��������
����ϸ������ ��ǻ��
����ϸ������
http://16629.dis.999120.net ��������
����ϸ���� ��ǻ��
����ϸ��������Һϸ���ٰ�����ϸ���ٰ�������ϸ������serouscelladenocarcinoma��aciniccelladenocarcinoma��aciniccelladenoma
http://16630.dis.999120.net ��������
�Һ��Ƥ���� ��ǻ��
�Һ��Ƥ����������
http://16631.dis.999120.net ��������
�������� ��ǻ��
����������Բ����
http://16632.dis.999120.net ��������
��ͷ״���ٰ� ��ǻ��
��ͷ״���ٰ��������Һ������ͷ��(�DZ�Ƥ��)��������ͷ״����������ͷ״�ٰ������ٰ���mucus-producingadenopapillary(nonepidermoid)carcinoma��malignantpapillarycystadenoma��papillaryadenocarcinoma��cystadenocarcinoma
http://16633.dis.999120.net ��������
���ٵ��ܰ� ��ǻ��
���ٵ��ܰ�
http://16634.dis.999120.net ��������
��ɫ����״���� ��ǻ��
��ɫ����״���룻���������ڰǻ�������ɫ�����������ף��ں�Ƥ�������װߣ���ǻ�Ĥ�����ʺ�ǻ���������ɫ�����Ĥ������whitefoldeddisease��leukokeratosisoris��whitefoldedgingivostomatitis��pachydermiaoris��softleukoplakia��naevus-likeisolatedhypertrophickera
http://16635.dis.999120.net ��������
��ǻ�Ĥ����ά�� ��ǻ��
��ǻ�Ĥ����ά��
http://16636.dis.999120.net ��������
�����Խǻ�����֢ ��ǻ��
�����Խǻ�����֢�������Խǻ������������Խǻ��쳣��Zinssers�ۺ���
http://16637.dis.999120.net ��������
��ɫˮ�� ��ǻ��
��ɫˮ��
http://16638.dis.999120.net ��������
����he ��ǻ��
����he
http://16639.dis.999120.net ��������
����ؽ������ۺ��� ��ǻ��
����ؽ������ۺ���������ؽ�����֢������ؽڹ��������ۺ�����temporomandibularjointdisorderssyndrome
http://16640.dis.999120.net ��������
���Դ��� ��ǻ��
���Է������Դ��ף����Դ���
http://16641.dis.999120.net ��������
���Դ��� ��ǻ��
���Դ���
http://16642.dis.999120.net ��������
�����ܰ���֯�����Դ��� ��ǻ��
�����ܰ���֯�����Դ��ף��ܰ������Դ��ף������ܰ���֯������cheilitisofbenignlymphoplasia
http://16643.dis.999120.net ��������
��ϸ���Դ��� ��ǻ��
��ϸ���Դ���
http://16644.dis.999120.net ��������
��ѿ���Դ��� ��ǻ��
��ѿ���Դ��ף��ʴ��Դ��ף�����hypertrophiccheilitis��macrocheilia
http://16645.dis.999120.net ��������
÷-���ۺ��� ��ǻ��
÷-���ۺ�������ѿ���Դ����ۺ��������ס���̱����������֢��
http://16646.dis.999120.net ��������
�չ��Դ��� ��ǻ��
�չ��Դ��ף��⻯�Դ��ף�actiniccheilitis
http://16647.dis.999120.net ��������
��̬��Ӧ�Դ��� ��ǻ��
��̬��Ӧ�Դ��ף���Ѫ������ˮ�ף��Ӵ��Դ���
http://16648.dis.999120.net ��������
Ӫ�������Կڽ��� ��ǻ��
Ӫ�������Կڽ��ף�Ӫ�������Կڽ���
http://16649.dis.999120.net ��������
����ؽڼ���ǰ��λ ��ǻ��
����ؽڼ���ǰ��λ
http://16650.dis.999120.net ��������
��ؽڸ�������λ ��ǻ��
��ؽڸ�������λ
http://16652.dis.999120.net ��������
��ؽڳ¾�����λ ��ǻ��
��ؽڳ¾�����λ
http://16654.dis.999120.net ��������
��ͼ�� ��ǻ��
���������ף���ͼ�ࣻgeographicglossitis
http://16656.dis.999120.net ��������
����ؽ���ǿֱ ��ǻ��
����ؽ���ǿֱ����������ؽ�ǿֱ
http://16657.dis.999120.net ��������
������ ��ǻ��
�ѹ��ࣻ�����ࣻ�����ࣻ�����ࣻ�����ࣻfissuredtongue��rugaetongue��linguaplicata��scrotaltongue
http://16658.dis.999120.net ��������
����ؽ���ǿֱ ��ǻ��
����ؽ���ǿֱ�����Թؽ�ǿֱ��������
http://16659.dis.999120.net ��������
���������ؽ�ǿֱ ��ǻ��
���������ؽ�ǿֱ
http://16660.dis.999120.net ��������
����ͷ�� ��ǻ��
����ͷ��
http://16661.dis.999120.net ��������
ë�� ��ǻ��
�
http://16662.dis.999120.net ��������
������������ ��ǻ��
������������
http://16663.dis.999120.net ��������
�������ʴ� ��ǻ��
�������ʴ����������
http://16664.dis.999120.net ��������
����������� ��ǻ��
����������ԣ�amyloidosislingualis
http://16665.dis.999120.net ��������
ή�������� ��ǻ��
ή�������ף���ɫƽ���ࣻ������
http://16666.dis.999120.net ��������
�he ��ǻ��
�he
http://16667.dis.999120.net ��������
Horner�ۺ��� ��ǻ��
������������ۺ�����Horner�ۺ�����Bernard-Horner�ۺ���
http://16668.dis.999120.net ��������
��he ��ǻ��
��he
http://16669.dis.999120.net ��������
���� ��ǻ��
����
http://16670.dis.999120.net ��������
���� ��ǻ��
����
http://16671.dis.999120.net ��������
���� ��ǻ��
����

http://16672.dis.999120.net ��������
�������� ��ǻ��
��������
http://16673.dis.999120.net ��������
���� ����
���ˣ�����
http://16675.dis.999120.net ��������
ͷƤ�������� ����
ͷƤ��������
http://16676.dis.999120.net ��������
���沿���� ����
���沿����
http://16677.dis.999120.net ��������
������ ����
������
http://16678.dis.999120.net ��������
������ ����
������
http://16679.dis.999120.net ��������
������ ����
������
http://16680.dis.999120.net ��������
�ǹؽ����� ����
�ǹؽ�����
http://16681.dis.999120.net ��������
���������� ����
����������
http://16682.dis.999120.net ��������
���������� ����
����������
http://16683.dis.999120.net ��������
���˺��Ĺ��ܲ�ȫ ����
���˺��Ĺ��ܲ�ȫ
http://16684.dis.999120.net ��������
���˺��Ժ��������ۺ��� ����
���˺��Ժ��������ۺ��������˺��Է����ˣ����˺��Ժ��������ۺ�֢
http://16685.dis.999120.net ��������
���˺�β���Ⱦ ����
���˺�β���Ⱦ
http://16686.dis.999120.net ��������
���˺��ˮ�� ����
���˺��ˮ��
http://16687.dis.999120.net ��������
���˺��˨�� ����
���˺��˨��
http://16688.dis.999120.net ��������
���˺��������ܲ�ȫ ����
���˺��������ܲ�ȫ
http://16690.dis.999120.net ��������
���˺������������� ����
���˺�������������
http://16691.dis.999120.net ��������
���˺�ϵĤ�϶����ۺ��� ����
���˺�ϵĤ�϶����ۺ��������˺�ϵĤ�϶����ۺ�֢
http://16692.dis.999120.net ��������
���˺�ι��ܲ�ȫ ����
���˺�ι��ܲ�ȫ
http://16693.dis.999120.net ��������
���˺���ʯ�Ե����� ����
���˺���ʯ�Ե�����
http://16694.dis.999120.net ��������
���˺��������� ����
���˺���������
http://16695.dis.999120.net ��������
���˺��� ����
���˺���
http://16697.dis.999120.net ��������
�����ݿ� ����
�����ݿˣ����������ݿ�
http://16698.dis.999120.net ��������
���˸�Ⱦ ����
���˸�Ⱦ
http://16699.dis.999120.net ��������
�������� ����
��������
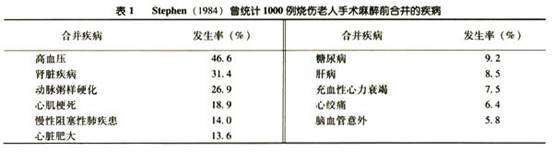
http://16700.dis.999120.net ��������
������ ����
������
http://16701.dis.999120.net ��������
��ѧ���� ����
��ѧ����
http://16702.dis.999120.net ��������
���������� ����
����������
http://16703.dis.999120.net ��������
��˹��ը�� ����
��˹��ը�ˣ�burnduetogasexplosion
http://16704.dis.999120.net ��������
������ ����
������

http://16705.dis.999120.net ��������
�����´� �ۿ�
�����´�
http://16706.dis.999120.net ��������
���ڷ� �ۿ�
���ڷ��������ڷ�
http://16707.dis.999120.net ��������
���ⷭ �ۿ�
���ⷭ�������ⷭ
http://16708.dis.999120.net ��������
���������� �ۿ�
���������ף�������
http://16709.dis.999120.net ��������
������ �ۿ�
�����ף������ף�������
http://16710.dis.999120.net ��������
����Ĥ������Χ�� �ۿ�
����Ĥ������Χ�ף�Eales�������긴���Բ������Ѫ��Ealesdisease
http://16711.dis.999120.net ��������
�������� �ڷ��ڿ�
�����������ȵ�������������
http://16713.dis.999120.net ��������
������ ����
������
http://16714.dis.999120.net ��������
������ ������
������
�������Ǻ�������������ͣ���Ϊ�ֻ������������ʽ�������������ʹ������������̬����Ҳ����ˡ� ����������������������صĽ��������������ȣ��������������������ܰ������dz�ϡ�٣�������ڲ�����ٷ���ת�ƣ���Σ������������������ͨ�����ڵĵ���Ĥ��ɢ���ּ��������������Լ�϶���������������϶������;���� �������ڷ������ڼ�����֢״����Ϊ������������仯�������������ĸı䡣�������������ܰͽ��״�����߲�������ܰ�ת�Ʒ��������ڲ��䡣
�������Ǻ�������������ͣ���Ϊ�ֻ������������ʽ�������������ʹ������������̬����Ҳ����ˡ����������������ǰ2/3,С���ַ�����ǰ���ϣ����ٷ����ں����ϡ�
http://16715.dis.999120.net ��������
������� ����
�������
http://16716.dis.999120.net ��������
�������� ����
��������
������28��ǰ��ֹ�߳�Ϊ��������������12��ǰ��Ȼ��ֹ�߳�����������������13?27����Ȼ��ֹ��Ϊ�����������Ӳ�ͬ��������ͬ�ײ㼰��ͬ�����ͳ�ƣ���Ȼ�����ķ�������15%?40%��Լ75%����������16����ǰ������������12��ǰ��ռ62%��
�����ӿ�ʼ��չ���սᾭ��һϵ�й��̣������䲻ͬ�ĽΣ��ɸ��費ͬ��������ƣ��ֱ�Ϊ������������������������ȫ��������ȫ����������������
http://16717.dis.999120.net ��������
�������� ����
��������
����ڱ�̥�Ĺ�����������ѪԽ��Խ�࣬�ﵽ����ƽʱ���¾����������۵�ҲԽ��Խ�����ˣ���ͱ��������ѽ��뵽���ɱ���Ľ��ˡ����������������ʱ��̥�Ѳ������ˡ��ٽ�һ����չ�����ܻ���һ������֯�����ۺ�ɫ����һ���Ķ������������ų���
һ����������Ϊ�����������ţ�����֯���������ˮ���������̥Ĥ��������ӹ���С��ͣ���������ϻ�ƫСʱ���Ϳ������Ϊ����������
http://16718.dis.999120.net ��������
��ȫ���� ����
��ȫ����
http://16719.dis.999120.net ��������
�������� ����
��������;��������
http://16720.dis.999120.net ��������
����ȫ���� ����
����ȫ����
http://16721.dis.999120.net ��������
��Ⱦ������ ����
��Ⱦ������
http://16722.dis.999120.net ��������
ϰ�������� ����
ϰ��������
ϰ��������������Ϊ�ӹ����ڿ��ɳ����¡������ڹι������Ź�����������ӹ��������ˣ������������������Է����쳣�����ಡ������������֮��������ˮ������̥������ǻ��ѹ�����ߣ�̥�ҿ��Թ����ڿ�ͻ��������ǻ��ѹ��������һ���̶ȣ��ͻ���Ĥ��������������ǰ����û���Ծ�֢״��
http://16723.dis.999120.net ��������
������� ����
�������
ȷ��Ϊ�������������:��1��������28?37��;��2��̥Ĥ����;��3�����ڿ���3cm����; ��4���ӹ���������ʱ�䲻����30s����϶Ϊ10min�����ߡ�
http://16724.dis.999120.net ��������
����ϲ����ಡ ����
����ϲ����ಡ
http://16725.dis.999120.net ��������
����ʧ�����ӹ���Ѫ ����
����ʧ�����ӹ���Ѫ
http://16726.dis.999120.net ��������
ԭ���Ավ� ����
ԭ���Ավ�
http://16727.dis.999120.net ��������
�̷��Ավ� ����
�̷��Ավ�
http://16728.dis.999120.net ��������
�ӹ����� ����
�ӹ�����
http://16729.dis.999120.net ��������
��ʴ������̥ ����
��ʴ������̥
http://16730.dis.999120.net ��������
����̥ ����
����̥����������̥
http://16731.dis.999120.net ��������
��ȫ����̥ ����
��ȫ����̥;��ȫ������̥
http://16732.dis.999120.net ��������
��ëĤ�� ����
��ëĤ��
http://16733.dis.999120.net ��������
������ ����
������
��������ָ���ٵļ��Ի�ŧ�Ը�Ⱦ���Dz����ڵij�����������������ȵ�ԭ��֮һ������ڲ��鸾Ů�������dz������������ڵ��κ�ʱ����ɷ�����������Ŀ�ʼ��Ϊ������
�������dz�����������һ�ֲ�֢�����߲��ܸ�Ӥ������ι�̣�������Ҫ�������ơ����ܼ���Ԥ�����ֺ�ʱ���ƣ��ɱ������Ს֢����ǰÿ������ͷ�������ϲ�һ�λ����ͣ�����8���º�ÿ���þƾ�����ˮϴ����ͷ�����Σ�ʹ��ͷƤ��������ĥ��Ԥ������Ӥ����˱�����ѡ�
http://16734.dis.999120.net ��������
С�����Ժ��� ����
http://16735.dis.999120.net ��������
��ͯ˯���ϰ� ����
��ͯ˯���ϰ���dyssomnia��sleepingdisturbance��somnipathy������״̬
http://16736.dis.999120.net ��������
���ȸ�ð ����
���ȸ�ð
http://16737.dis.999120.net ��������
������������ ����
������������
http://16738.dis.999120.net ��������
����������÷�� ����
����������÷��
http://16739.dis.999120.net ��������
�����Ը��ű��� ����
���ء�����֢
http://16740.dis.999120.net ��������
����ʹ ����
����ʹ
http://16741.dis.999120.net ��������
�������������Բ� ����
�������ж����Բ�
http://16742.dis.999120.net ��������
���ܰͽ��� ����
���ܰͽ���
http://16743.dis.999120.net ��������
ëϸѪ���� ����
Ӥ����Ѫ����
http://16744.dis.999120.net ��������
����˹-��̫˹�� Ƥ����
����˹-��̫˹��������˹-����˹��������˹-����˹���ϲ�������ë�ҽǻ�֢��Ҹ���������ں���(����)��
http://16745.dis.999120.net ��������
������ Ƥ����
��������eccrinesyringocystadenoma��eruptivahidroadenoma��syringocysadenoma��syringocystoma��syringo-hidroadenoma�������Ժ���������״�����������ܺ���������������������������������������С���ٺ�����